Spatial Pharmacology: Organelle Targeting and Condensate-Mediated Drug Discovery
Spatial Pharmacology: Organelle Targeting and Condensate-Mediated Drug Discovery
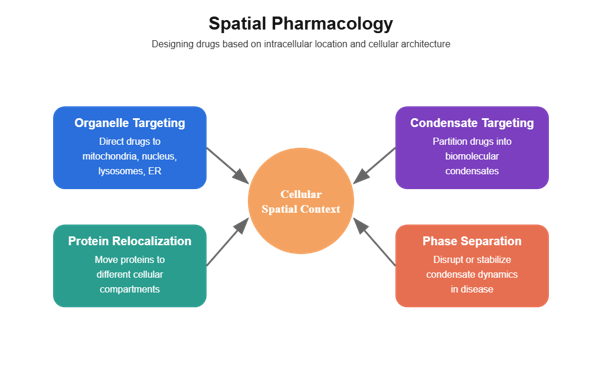
Figure: Conceptual overview of spatial pharmacology. Spatial pharmacology designs therapeutics based on intracellular location and cellular architecture. Organelle-targeted therapeutics direct drugs to specific compartments such as mitochondria or lysosomes. Protein relocalization strategies move proteins to alternative cellular locations to alter function. Condensate-targeting approaches exploit biomolecular phase separation to concentrate drugs within transcriptional or signaling condensates. Phase-separation therapeutics modulate the formation or stability of these condensates to influence disease pathways.
Executive Overview: From Molecular Binding to Spatial Control
For most of modern drug discovery, success has been defined by affinity, potency, and selectivity at the molecular level. A compound binds a protein, inhibits an enzyme, blocks a receptor. But cells are not homogeneous reaction vessels. They are spatially organized systems composed of:
- Membrane-bound organelles
- Dynamic trafficking networks
- Phase-separated condensates
- Compartment-specific microenvironments
A molecule’s biological effect depends not only on what it binds — but where it goes.
Spatial pharmacology represents a paradigm shift: therapeutics are designed with intracellular geography in mind. Instead of merely targeting a protein, drugs can:
- Relocalize proteins to different organelles
- Partition selectively into biomolecular condensates
- Exploit phase separation dynamics
- Engage bacterial or mitochondrial proteostasis machinery
- Alter the spatial context of signaling networks
This emerging field encompasses:
- Organelle-targeted therapeutics
- Protein relocalization strategies
- Condensate-mediated drug discovery
- Phase separation therapeutics
- Spatially restricted proteolysis systems
As our understanding of cellular architecture deepens, spatial control is becoming a new axis of drug design.
The Rise of Spatial Pharmacology
Why Location Matters in Biology
Cells compartmentalize reactions to:
- Increase efficiency
- Prevent crosstalk
- Protect genomic integrity
- Regulate signaling thresholds
Key compartments include:
- Nucleus
- Mitochondria
- Endoplasmic reticulum
- Lysosomes
- Endosomes
- Peroxisomes
Additionally, cells contain membraneless organelles, formed via liquid–liquid phase separation (LLPS). These condensates concentrate proteins and RNA into dynamic microdomains.
Drugs that ignore this spatial complexity may:
- Fail to reach their intended microenvironment
- Be sequestered away from targets
- Disrupt unintended compartments
Spatial pharmacology embraces compartmentalization as a design principle rather than a pharmacokinetic obstacle.
Organelle-Targeted Therapeutics
Organelle-targeted therapeutics aim to direct molecules — or target proteins themselves — to specific intracellular locations.
Direct Organelle Targeting
Certain chemical motifs preferentially accumulate in specific organelles. For example:
- Lipophilic cations accumulate in mitochondria
- Acidic compartments trap weak bases
- Nuclear localization sequences direct nuclear import
By engineering compounds with organelle-targeting elements, researchers can:
- Enhance local concentration
- Improve target engagement
- Reduce systemic toxicity
Targeted Protein Relocalization
Beyond delivering drugs to organelles, another strategy is to move proteins themselves.
Conceptual Framework
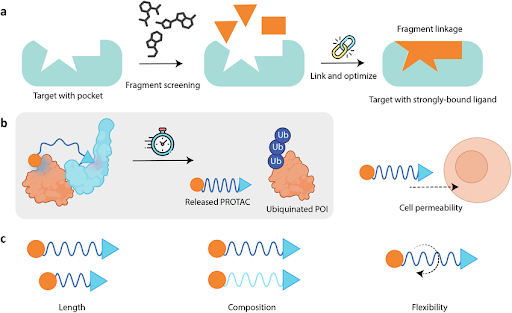
Small molecules or bifunctional chimeras can bind a protein of interest and tether it to:
- A mitochondrial membrane
- A nuclear export receptor
- A lysosomal surface
- A cytoskeletal anchor
Relocalization can:
- Inactivate signaling pathways
- Prevent nuclear transcriptional activity
- Induce degradation through organelle-associated mechanisms
- Alter metabolic flux
This represents functional knockdown without necessarily degrading the protein.
Mitochondrial Targeting as a Case Study
Mitochondria are central to:
- Energy metabolism
- Apoptosis regulation
- Reactive oxygen species production
Organelle-targeted therapeutics in oncology may:
- Induce mitochondrial stress
- Sensitize cells to apoptosis
- Exploit tumor metabolic rewiring
Conversely, in neurodegeneration, mitochondrial targeting can:
- Reduce oxidative stress
- Restore bioenergetic balance
Spatial precision improves therapeutic index.
Condensate-Mediated Drug Discovery
What Are Biomolecular Condensates?
Condensates are membraneless compartments formed through liquid–liquid phase separation (LLPS). They arise when multivalent interactions among proteins and RNA cause demixing from the surrounding cytoplasm or nucleoplasm.
Examples include:
- Nucleoli
- Stress granules
- P-bodies
- Transcriptional super-enhancer condensates
- DNA damage foci
These structures concentrate specific biomolecules while excluding others.
Why Condensates Matter for Drug Discovery
Condensates influence:
- Gene transcription
- RNA processing
- Signal transduction
- Stress responses
- DNA repair
Drugs can:
- Partition preferentially into condensates
- Alter phase separation dynamics
- Disrupt scaffold-client interactions
- Stabilize or dissolve condensates
Thus, drug behavior is not solely defined by binding affinity — but by partitioning behavior within mesoscale cellular structures.
Drug Partitioning into Condensates
Certain small molecules concentrate within condensates due to:
- Electrostatic interactions
- π–π stacking
- Intrinsically disordered region (IDR) affinity
- RNA binding properties
Partitioning can amplify local drug concentration beyond cytoplasmic levels. However, it can also:
- Sequester drugs away from intended targets
- Create off-target effects within transcriptional hubs
Understanding condensate partition coefficients is becoming a critical parameter in medicinal chemistry.
Modulating Phase Separation as Therapy
Phase separation therapeutics aim to:
- Dissolve oncogenic transcriptional condensates
- Stabilize protective condensates
- Prevent pathological aggregation
In oncology, super-enhancer condensates regulate oncogene expression. Disrupting their integrity can suppress transcriptional addiction.
In neurodegeneration, aberrant phase transitions may drive toxic aggregation. Modulating condensate fluidity could prevent pathological conversion.
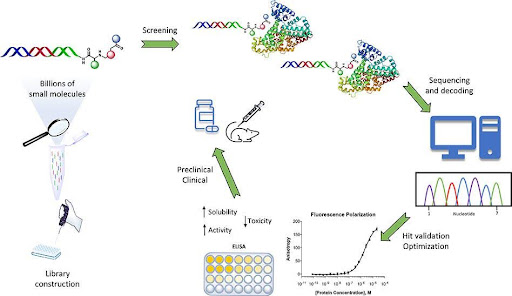
Screening Strategies in Condensate Drug Discovery
Modern screening approaches include:
- In vitro reconstitution of phase separation
- High-content live-cell imaging
- Fluorescence recovery after photobleaching (FRAP)
- Quantitative partitioning assays
Drug discovery is evolving from simple binding assays to mesoscale biophysical profiling.
Spatial Proteostasis and Bacterial Systems
BacPROTACs: Spatial Targeting in Bacteria
BacPROTACs adapt induced proximity logic to bacterial proteostasis machinery. Rather than recruiting human E3 ligases, BacPROTACs engage bacterial proteases such as Clp systems.
Implications
- Novel antibacterial strategies
- Targeted degradation of resistance factors
- Reduced reliance on enzyme inhibition
Bacterial cells lack compartmental complexity comparable to eukaryotes, but spatial organization of proteases still governs degradation efficiency.
Designing Drugs with Spatial Awareness
Subcellular Distribution Profiling
Beyond plasma concentration, developers must assess:
- Nuclear accumulation
- Mitochondrial enrichment
- Lysosomal sequestration
- Condensate partitioning
Advanced imaging techniques now quantify intracellular distribution at high resolution.
Chemical Properties and Compartment Bias
Physicochemical parameters influence spatial behavior:
- Charge
- Lipophilicity
- Polar surface area
- Hydrogen bonding capacity
Medicinal chemists must balance permeability with compartment selectivity.
Temporal Dynamics
Spatial localization is dynamic. Stress, cell cycle stage, and signaling events alter compartment architecture.
A drug may behave differently in:
- Hypoxic tumors
- Neurons vs hepatocytes
- Inflamed tissues
Time-resolved spatial profiling becomes essential.
Disease Applications of Spatial Pharmacology
Oncology
Cancer cells often reorganize spatial architecture:
- Super-enhancer condensates drive oncogene transcription
- DNA repair condensates maintain genomic instability
- Metabolic enzymes relocalize under stress
Spatial pharmacology strategies may:
- Disrupt transcriptional condensates
- Relocalize oncogenic proteins
- Target mitochondrial vulnerabilities
Neurodegeneration
Many neurodegenerative diseases involve:
- Protein aggregation
- Stress granule dysfunction
- Aberrant phase transitions
Condensate drug discovery may prevent pathological solidification of liquid compartments.
Organelle-targeted therapeutics may protect:
- Mitochondria
- Lysosomes
- Synaptic compartments
Infectious Disease
Pathogens manipulate host spatial organization:
- Viral replication factories
- Bacterial inclusion bodies
Spatially aware drugs could disrupt these specialized compartments.
Metabolic and Rare Diseases
Organelle dysfunction underlies:
- Mitochondrial disorders
- Lysosomal storage diseases
- Peroxisomal disorders
Targeted delivery to affected organelles may improve therapeutic efficacy.
Resistance and Adaptive Remodeling
Cells adapt spatial architecture under drug pressure. Potential resistance mechanisms include:
- Condensate remodeling
- Organelle biogenesis changes
- Compensatory trafficking pathways
- Altered partitioning behavior
Understanding spatial plasticity is critical for durable therapy.
Integration with Other Modalities
Spatial pharmacology intersects with:
- Targeted protein degradation
- Synthetic lethality
- RNA-targeted therapeutics
- Immunotherapy
For example:
- Degraders may preferentially act within condensates
- Synthetic lethal vulnerabilities may arise from organelle stress
- RNA-targeted drugs may accumulate in nuclear condensates
Future therapeutics may combine spatial targeting with induced proximity.
The Role of AI and Computational Modeling
Advances in machine learning enable:
- Prediction of phase separation propensity
- Modeling of drug partition coefficients
- Simulation of organelle localization
- Integration of multi-omic spatial datasets
Computational spatial pharmacology may soon guide medicinal chemistry from the earliest stages.
The Commercial Landscape
Biotech innovation in spatial pharmacology is accelerating. Emerging companies focus on:
- Condensate-disrupting compounds
- Organelle-targeted degraders
- Spatially restricted proteostasis modulation
- Mesoscale screening platforms
Pharmaceutical interest is rising because:
- Spatial targeting expands the druggable proteome
- Novel mechanisms create differentiation
- Biomolecular condensates represent underexploited targets
Future Directions in Spatial Pharmacology
The next frontier may include:
- Multi-compartment targeting chimeras
- Programmable relocalization systems
- Condensate-selective degraders
- Organelle-specific E3 ligase atlases
- Spatially resolved precision oncology
As spatial multi-omics advances, vulnerability maps may include not just gene expression — but subcellular context.
Conclusion: The Third Dimension of Drug Discovery
Traditional pharmacology operates in two dimensions:
- Molecular affinity
- Target selectivity
Spatial pharmacology introduces the third:
- Intracellular location
Organelle-targeted therapeutics and condensate-mediated drug discovery redefine what it means to engage a target. The effectiveness of a drug may depend not only on binding strength, but on its ability to navigate cellular geography.
By integrating knowledge of:
- Organelle biology
- Phase separation
- Intracellular trafficking
- Mesoscale organization
drug discovery becomes not just chemistry and biology — but cellular architecture engineering.
The future of medicine may hinge on mastering this spatial dimension. In the coming decade, spatial pharmacology is poised to transform how we conceptualize therapeutic intervention — from blocking molecular function to orchestrating cellular organization itself.
Related Services
| Service | |
|---|---|
| Small molecule drug discovery for even hard-to-drug targets – identify inhibitors, binders and modulators | |
| Molecular Glue Direct | |
| PPI Inhibitor Direct | |
| Integral membrane proteins | |
| Specificity Direct – multiplexed screening of target and anti-targets | |
| Express – optimized for fast turn – around-time | |
| Snap – easy, fast, and affordable |