


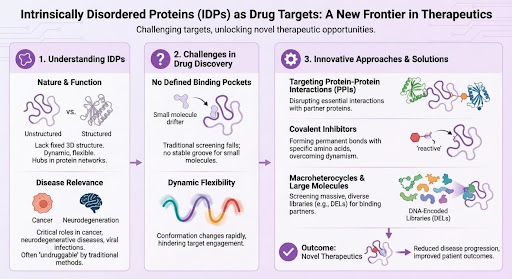
Intrinsically Disordered Proteins as Drug Targets: Challenges, Opportunities, and Screening Strategies

For decades, drug discovery has been most comfortable with proteins that behave like engineered locks: stable, well folded, and furnished with pockets that make it relatively straightforward to design a key. Intrinsically disordered proteins, or IDPs, do not behave that way. Instead of occupying a single rigid structure, they exist as dynamic ensembles of interconverting conformations. That structural plasticity is exactly what makes them so important in biology, and exactly what has made them so frustrating in medicinal chemistry [1–4].
Yet the old view that intrinsically disordered proteins are simply “undruggable” is becoming outdated. The modern question is no longer whether disorder can be targeted at all, but how to match the right discovery strategy to the biology of the target. In many cases, IDPs are deeply embedded in disease-relevant pathways involving transcriptional control, protein-protein interactions, condensate formation, and aggregation. Those are all mechanisms that matter in oncology, neurodegeneration, inflammation, and other high-value therapeutic areas [1,4].
That shift in perspective is especially relevant for companies working on hard-to-drug targets. If an intrinsically disordered region drives function through transient contacts, weak multivalent interactions, or context-dependent conformational changes, the challenge is not only one of medicinal chemistry. It is also a screening problem. The discovery engine must be capable of finding binders and modulators against targets that may not present a classical active site. This is where technologies such as DNA-encoded library, or DEL, screening become highly relevant to the IDP field [18–22].
What are intrinsically disordered proteins?
Intrinsically disordered proteins are proteins, or protein regions, that do not adopt a single stable three-dimensional structure under physiological conditions [1]. Rather than being misfolded or nonfunctional, they often remain disordered because that flexibility is biologically useful. Intrinsically disordered regions (IDRs) can act as hubs for signaling, regulation, assembly, post-translational modification, and selective partner recognition. Their conformational heterogeneity lets them bind multiple partners, switch states, and respond rapidly to changes in the cellular environment [1,2].
This “disorder-function” relationship is especially important in eukaryotic biology. Many regulatory proteins rely on disorder to integrate inputs and coordinate outputs. IDPs are therefore common in transcriptional machinery, signaling complexes, cell-cycle control, RNA regulation, and stress responses [1,3]. In practical drug discovery terms, that means some of the most biologically compelling targets are also among the least structurally well behaved.
A useful way to think about IDPs is that they are not structureless; they are structurally distributed. They populate ensembles rather than single conformations. Some regions may transiently sample helices, turns, or compact states. Others may become more ordered only upon interaction with a partner. This matters for ligand discovery because the target is not a fixed object. It is a moving landscape, and small molecules may act not by occupying a permanent pocket, but by shifting populations within that landscape [4–6].
Why intrinsically disordered proteins matter in disease
IDPs are not just biochemical curiosities. They are overrepresented in pathways that sit close to disease causality. Because they are often involved in signaling and transcriptional control, even subtle dysregulation can produce large phenotypic consequences. That is one reason disordered proteins and regions repeatedly appear in cancer biology, neurodegenerative disease, inflammatory signaling, viral biology, and autoimmune disorders [1,4].
Cancer is the most obvious example. Many transcription factors, co-regulators, and chromatin-associated proteins contain substantial intrinsically disordered regions. Those regions can mediate cofactor recruitment, DNA-context sensing, condensate behavior, and dynamic protein-protein interactions that are essential for oncogenic gene expression programs [7,8]. Similarly, in neurodegeneration, disordered proteins and low-complexity regions are central to aggregation and phase-transition phenomena that contribute to pathology [4,11,15].
These targets are attractive precisely because they are functionally central. But their importance also means that conventional discovery approaches can miss them. When a target’s key biology depends on transient interaction surfaces, disorder-to-order transitions, or phase-separated assemblies, methods optimized for rigid enzymes can underperform. The implication is strategic: hard target biology requires hard target screening logic.
Why IDPs were long considered “undruggable”
The traditional medicinal chemistry playbook was built around stable pockets, often in enzymes or receptors. By comparison, many IDPs appear to lack deep, persistent cavities. Their interfaces can be broad, shallow, and highly dynamic. In addition, binding may depend on a specific cellular context, a partner protein, a post-translationally modified state, or a multicomponent assembly. All of that makes structure-guided design much harder [2–6].
Several technical barriers follow from that biology. First, structural characterization is more difficult. Disordered proteins often require integrative methods such as NMR, SAXS, hydrogen-deuterium exchange, single-molecule approaches, or ensemble modeling rather than conventional crystallography alone [4,6]. Second, assay design becomes more delicate. Measuring binding, selectivity, and mechanism is more challenging when the target samples many conformations and when function may depend on weak, multivalent interactions rather than a single binary event [6].
Third, the desired pharmacology is often indirect. For an enzyme, one may want active-site occupancy. For an IDP, one may instead want to destabilize a pathological interaction, bias the conformational ensemble, alter condensate partitioning, prevent aggregation, or block recruitment of a crucial partner [4,6,11]. Once that shift is understood, “undruggable” starts to look less like a property of the target and more like a limitation of the discovery framework used against it.
Why intrinsically disordered proteins are increasingly considered druggable
A major conceptual advance in the field is the recognition that disorder does not eliminate ligandability. It changes ligandability. Instead of asking whether a protein has a single textbook pocket, researchers increasingly ask whether the conformational ensemble contains transiently populated cavities, inducible interaction surfaces, or energy minima that can be shifted by a ligand [4–6]. Computational and biophysical studies support this view, showing that disordered proteins can present transient cavities and druggable microstates even when no rigid pocket is evident in a static model [5,16].
This ensemble-based view is important because it expands the range of possible mechanisms. A small molecule can bind a subset of conformations. It can stabilize a less functional state. It can compete with a partner at a transient interface. It can alter the kinetics of folding-upon-binding. It can change the material properties of a condensate. Or it can suppress pathogenic self-association without needing an enzyme-like pocket [4–6,11].
Recent literature frames this as one of the next major hurdles in drug discovery rather than a dead end. The field is moving toward integrated workflows that combine computation, biophysics, orthogonal screening, and context-aware assays to identify compounds that act on disorder-mediated biology [4,6,16]. That framing is highly relevant for industrial discovery programs because it argues against a one-method solution. IDPs generally reward multimodal hit finding and careful follow-up.
Transcription factors, PPIs, and other hard-to-drug targets are often IDP problems
One reason the IDP topic matters commercially is that it overlaps so strongly with classes already recognized as difficult targets. Transcription factors are a prime example. Many transcription factors contain intrinsically disordered regions that contribute to cofactor recruitment, DNA-binding specificity, responsiveness to post-translational modification, and formation of dynamic transcriptional assemblies [7,8]. That means a campaign against a transcription factor often becomes, at least in part, a campaign against protein disorder.
Protein-protein interactions create a similar challenge. Broad, shallow, adaptable interfaces are common in disorder-mediated recognition. In some cases, disordered segments fold only when they meet the right partner. In others, the interaction remains fuzzy or partially heterogeneous even in the bound state. Either way, the ligand discovery problem is fundamentally different from screening a stable catalytic domain [2,6,7].
This is one reason transcription factors have moved from the “undruggable” bucket into the “difficult but actionable” bucket. The literature now contains multiple examples of successful or at least credible targeting strategies, including direct binding to disordered regions, disruption of partner interactions, and modulation of transcriptional complexes [7].
Biomolecular condensates add a new dimension to IDP drug discovery
The IDP story has broadened further with the rise of biomolecular condensates and liquid-liquid phase separation. Many condensates are enriched in disordered and low-complexity regions that support multivalent interactions, dynamic assembly, and selective partitioning of proteins and nucleic acids [9–11]. For drug discovery, this matters because the relevant biology may not be a single target-ligand interaction in dilute solution. It may be a target embedded in a mesoscale assembly with different local concentration, dynamics, and composition.
Condensate biology also expands the types of pharmacology that may be useful. A compound might reduce or enhance phase separation, alter condensate viscosity, bias recruitment of a partner, or change the balance between functional condensation and pathological aggregation [10,11]. This has obvious implications for oncology and neurodegeneration, where transcriptional condensates, stress granules, and aggregation-prone assemblies are active areas of therapeutic interest.
Importantly, condensates do not make small-molecule discovery impossible. They make context more important. A ligand that appears weak in a minimal biochemical setup may become highly relevant if it changes partitioning or assembly behavior in a more native system. Conversely, a strong binder in vitro may not translate if it fails to engage the relevant target state in cells [10,11]. This is another reason why IDP programs benefit from screening and validation workflows that can accommodate both purified protein and more physiological formats.
Case studies show that IDP targeting is feasible
The strongest argument against the “undruggable” label is empirical. Over the past two decades, multiple groups have reported small molecules that bind directly or functionally to intrinsically disordered proteins or regions.
The c-Myc oncoprotein remains one of the best-known examples. The bHLHZip region of monomeric c-Myc is intrinsically disordered, yet small molecules such as 10058-F4 and 10074-G5 (Scheme 1) were shown to bind disordered c-Myc, disrupt Myc-Max function, and reveal that distinct ligandable sites can exist within a disordered segment [12,13]. These studies were important not because they solved all medicinal chemistry challenges around Myc, but because they demonstrated a principle: a dynamic ensemble can still be targeted by small molecules.
Case studies show that IDP targeting is feasible
The strongest argument against the “undruggable” label is empirical. Over the past two decades, multiple groups have reported small molecules that bind directly or functionally to intrinsically disordered proteins or regions.
The c-Myc oncoprotein remains one of the best-known examples. The bHLHZip region of monomeric c-Myc is intrinsically disordered, yet small molecules such as 10058-F4 and 10074-G5 (Scheme 1) were shown to bind disordered c-Myc, disrupt Myc-Max function, and reveal that distinct ligandable sites can exist within a disordered segment [12,13]. These studies were important not because they solved all medicinal chemistry challenges around Myc, but because they demonstrated a principle: a dynamic ensemble can still be targeted by small molecules.

Scheme 1: Structure of 10058-F4, 10074-G5, and YK-4-279
Another influential example is EWS-FLI1, an oncogenic fusion transcription factor central to Ewing sarcoma. YK-4-279 (Scheme 1) and related work helped establish that a small molecule could disrupt functionally critical interactions associated with this difficult target class [14]. Likewise, α-synuclein studies showed that small molecules can engage a disordered ensemble and modify aggregation-related behavior, reinforcing the idea that disorder can be addressed through ensemble modulation rather than classical pocket blockade [15].
More recently, computational and integrative biophysical strategies have been used to identify compounds that bind disordered regions of p53 and other targets [6,16]. In prostate cancer, the intrinsically disordered N-terminal domain of the androgen receptor has become a recognized drug discovery focus, especially because targeting that region offers a path beyond ligand-binding-domain resistance mechanisms [17]. None of these examples suggest that IDP discovery is easy. They do show, however, that it is possible, increasingly systematic, and highly relevant to serious therapeutic programs.
What successful screening of intrinsically disordered proteins requires
Because IDPs often fail the assumptions built into classical screening, successful programs usually begin with a broader definition of what counts as a hit. In an enzyme program, a hit may be an inhibitor with clear active-site engagement. In an IDP program, a hit may instead be a binder that shifts a conformational ensemble, weakens a pathological interaction, changes condensate behavior, or creates a tractable starting point for follow-up chemistry [4,6].
That has practical consequences for assay design. A strong campaign often uses multiple target constructs, multiple buffer conditions, and more than one readout. Biophysical methods may be needed early to confirm that a candidate truly engages the target rather than producing an assay artifact. Cell-based assays can become particularly important when function depends on partner proteins, compartmentalization, or condensate formation [6,21,22].
Orthogonal validation is critical. Because disorder-mediated binding can be subtle and context dependent, apparent hits should ideally be tested across complementary assays that distinguish direct engagement from nonspecific stickiness, aggregation artifacts, or reporter interference. This is not unique to IDPs, but the need is usually more acute [6,21]. It is a staged workflow that goes from binder discovery to mechanism refinement in progressively relevant systems [4,22].
Where DNA-encoded library screening fits into IDP drug discovery
DNA-encoded libraries have become an important part of the modern hit-identification toolbox because they allow pooled screening of very large numbers of compounds through DNA barcoding and affinity selection [18–20]. For difficult targets, that scale matters. When the target surface is transient, shallow, or unconventional, broader chemical sampling can materially improve the odds of finding a productive starting point.
That is especially relevant for intrinsically disordered proteins. IDP programs often benefit from a binder-first mindset: find molecules that interact with the target or target state, then determine whether they can be evolved into useful modulators. DEL screening fits well with that logic because it is fundamentally a ligand-discovery technology. It is not limited to targets with obvious catalytic sites, and modern DEL methods have expanded beyond simple immobilized-protein formats toward more varied selection strategies [18–20].
DEL should not be presented as a magic bullet for IDPs. It still depends on target quality, construct design, assay conditions, follow-up chemistry, and orthogonal confirmation. But the industrial literature is clear that encoded library technologies are valuable in expanding hit finding into more difficult target spaces, and that the broader hit-identification landscape increasingly relies on complementary methods for challenging targets with disordered regions, PPIs, or protein-DNA interfaces [19,21,22]. For IDP campaigns, DEL can therefore be a pragmatic way to widen chemical exploration at the earliest stage.
Why DEL can be especially useful for hard-to-drug and disorder-rich targets
There are three reasons DEL is particularly attractive in this context.
First, it expands chemical reach. Difficult targets often require looking beyond the chemistry represented in conventional screening decks. DEL technology was built precisely to interrogate much larger libraries efficiently [18–20].
Second, it supports a binder-centric strategy. That is valuable for intrinsically disordered proteins, where the first objective is often to establish whether the target ensemble can be productively engaged at all. A binder does not have to be a finished inhibitor to be strategically valuable. It can serve as a probe, a starting point for medicinal chemistry, a structural handle, or a route into assay development and mechanistic refinement [19,22].
Third, DEL integrates well with orthogonal workflows. The strongest industrial practice today is not to rely on a single hit-finding technology, but to combine complementary approaches based on target biology and practical constraints [21,22]. In that framework, DEL is not competing against every other modality; it is part of an integrated discovery strategy. For intrinsically disordered proteins, that approach often the difference between a scientifically interesting target and a tractable program.
The future of targeting intrinsically disordered proteins
The field is now moving from proof-of-concept to platform maturity. Better ensemble modeling, better biophysics, better cellular assays, and better integration of orthogonal hit-finding technologies are all helping convert IDP biology into tangible discovery programs [4,6,16,22]. That does not mean every intrinsically disordered protein will become easy to drug, but it does mean the category is no longer scientifically or commercially marginal.
In the coming years, some of the most interesting advances are likely to come from the intersection of disorder biology with transcriptional regulation, condensate pharmacology, targeted degradation, and integrated screening [4,10,11,21,22]. In other words, the IDP problem is not a side story in modern drug discovery. It sits near the center of where the field is going.
For teams evaluating difficult targets today, the practical conclusion is clear: intrinsically disordered proteins should no longer be excluded simply because they violate the assumptions of classical structure-based drug discovery. They should instead be approached with a discovery strategy built for dynamic targets. In that setting, DNA-encoded library screening, especially when paired with orthogonal validation and biologically relevant assay formats, can be an important part of the answer [18–25].
Conclusion
Intrinsically disordered proteins are challenging drug targets because their biology is dynamic, context dependent, and often mediated by broad interaction surfaces rather than classical pockets. But that same biology is what makes them so therapeutically important. IDPs sit at the heart of transcription, signaling, condensation, and aggregation processes that drive major diseases [1,4,11].
The discovery landscape has changed accordingly. Modern work on c-Myc, EWS-FLI1, α-synuclein, p53, and the androgen receptor shows that disorder can be targeted through direct binding, conformational modulation, interaction disruption, and other nonclassical mechanisms [12–17]. The industrial implication is that hard targets require flexible hit-identification strategies, integrated assay design, and technologies capable of finding starting points where conventional methods struggle [19,21,22].
That is why intrinsically disordered proteins are now better understood not as impossible targets, but as targets that demand the right screening strategy. For companies focused on hard-to-drug biology, that is an opportunity.
References
[1] Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signaling and regulation. Nat Rev Mol Cell Biol. (2015), 16, 18-29. doi.org/10.1038/nrm3920
[2] Metallo SJ. Intrinsically disordered proteins are potential drug targets. Curr Opin Chem Biol. (2010), 14(4), 481-488. https://doi.org/10.1016/j.cbpa.2010.06.169
[3] Uversky VN. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin Drug Discov. (2012), 7, 475-488. https://doi.org/10.1517/17460441.2012.686489
[4] Lazar T, Connor A, DeLisle CF, Burger V, Tompa P. Targeting protein disorder: the next hurdle in drug discovery. Nat Rev Drug Discov. (2025), 24(10), 743-763. doi.org/10.1038/s41573-025-01220-6
[5] Zhang Y, Cao H, Liu Z. Binding cavities and druggability of intrinsically disordered proteins. Protein Sci. (2015), 24(5), 688-705. https://doi.org/10.1002/pro.2641
[6] Chen J, Liu X, Chen J. Targeting intrinsically disordered proteins through dynamic interactions. Biomolecules. (2020), 10(5), 743. https://doi.org/10.3390/biom10050743
[7] Tsafou K, Tiwari PB, Forman-Kay JD, Metallo SJ, Toretsky JA. Targeting intrinsically disordered transcription factors: changing the paradigm. J Mol Biol. (2018), 430(16), 2321-2341. https://doi.org/10.1016/j.jmb.2018.04.008
[8] Brodsky S, Jana T, Barkai N. Order through disorder: the role of intrinsically disordered regions in transcription factor binding specificity. Curr Opin Struct Biol. (2021), 71, 110-115. https://doi.org/10.1016/j.sbi.2021.06.011
[9] Hyman AA, Weber CA, Jülicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. (2014), 30, 39-58. https://doi.org/10.1146/annurev-cellbio-100913-013325
[10] Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. (2017), 18(5), 285-298. doi.org/10.1038/nrm.2017.7
[11] Biesaga M, Frigolé-Vivas M, Salvatella X. Intrinsically disordered proteins and biomolecular condensates as drug targets. Curr Opin Chem Biol. (2021), 62, 90-100. https://doi.org/10.1016/j.cbpa.2021.02.009
[12] Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol. (2008), 15(11), 1149-1155. https://doi.org/10.1016/j.chembiol.2008.09.011
[13] Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc. (2009), 131(21), 7390-7401. https://doi.org/10.1021/ja900616b
[14] Barber-Rotenberg JS, Selvanathan SP, Kong Y, Erkizan HV, Snyder TM, Hong SP, et al. Single enantiomer of YK-4-279 demonstrates specificity in targeting the oncogene EWS-FLI1. Oncotarget. (2012), 3(2), 172-182. https://doi.org/10.18632/oncotarget.454
[15] Tóth G, Gardai SJ, Zago W, Bertoncini CW, Cremades N, Roy SL, et al. Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLoS One. (2014), 9(2), e87133. https://doi.org/10.1371/journal.pone.0087133
[16] Ruan H, Yu C, Niu X, Zhang W, Liu H, Chen L, Xiong R, Sun Q, Jin C, Liu Y, Lai L. Computational strategy for intrinsically disordered protein ligand design leads to the discovery of p53 transactivation domain I binding compounds that activate the p53 pathway. Chem Sci. (2021), 12, 3004-3016. https://doi.org/10.1039/D0SC04670A
[17] Sadar MD. Discovery of drugs that directly target the intrinsically disordered region of the androgen receptor. Expert Opin Drug Discov. (2020), 15(5), 551-560. https://doi.org/10.1080/17460441.2020.1732920
[18] Kunig V, Potowski M, Gohla A, Brunschweiger A. DNA-encoded libraries – an efficient small molecule discovery technology for the biomedical sciences. Biol Chem. (2018), 399(7), 691-710. https://doi.org/10.1515/hsz-2018-0119
[19] Ottl J, Leder L, Schaefer JV, Dumelin CE. Encoded library technologies as integrated lead finding platforms for drug discovery. Molecules. (2019), 24(8), 1629. https://doi.org/10.3390/molecules24081629
[20] Huang Y, Li X. Recent advances on the selection methods of DNA-encoded libraries. ChemBioChem. (2021), 22(14), 2384-2397. https://doi.org/10.1002/cbic.202100144
[21] Lanne A, Usselmann LEJ, Llowarch P, Michaelides IN, Fillmore M, Holdgate GA. A perspective on the changing landscape of HTS. Drug Discov Today. (2023), 28(8), 103670. https://doi.org/10.1016/j.drudis.2023.103670
[22] Ashraf SN, Blackwell JH, Holdgate GA, Lucas SCC, Solovyeva A, Storer RI, Whitehurst BC. Hit me with your best shot: integrated hit discovery for the next generation of drug targets. Drug Discov Today. (2024), 29(10), 104143. https://doi.org/10.1016/j.drudis.2024.104143
[23] Vipergen. DNA-encoded library (DEL). Vipergen website. Accessed March 26, 2026. Link
[24] Vipergen. Discovery of inhibitors, binders and modulators. Vipergen website. Accessed March 26, 2026. Link
[25] Vipergen. Transcription Factor Inhibitors: From “Undruggable” to Drug Discovery Reality. Vipergen website. Accessed March 26, 2026. Link