



Enzymes remain one of the most important and consistently productive classes of drug targets in modern small-molecule discovery. Across oncology, immunology, infectious disease, neuroscience, inflammation, and metabolic disease, enzymes sit at mechanistically powerful control points: they catalyze pathway-defining reactions, shape signal transduction, regulate epigenetic states, process proteins, repair DNA, and control the concentrations of key metabolites. When those functions become dysregulated, selective chemical modulation can generate direct and therapeutically meaningful biological effects. This is one reason enzymes continue to occupy such a large share of successful drug discovery programs and marketed medicines [1,2].
From a discovery standpoint, enzymes are attractive because they are often intrinsically ligandable. They contain catalytic pockets, substrate-recognition elements, cofactor-binding sites, conformational switches, and, in many cases, allosteric pockets that can be addressed with small molecules. That combination makes them especially well suited to small molecule enzyme inhibitors, whether the goal is direct active-site blockade, allosteric modulation, or covalent engagement of a catalytic or nearby nucleophilic residue [1,3-8]. For this reason, kinase inhibitor discovery, protease inhibitor screening, and broader enzyme inhibitor discovery services remain central parts of early pharmaceutical R&D.
At the same time, enzyme drug discovery is rarely as simple as “find a potent inhibitor and move on.” Many enzyme families have highly conserved active sites. Others are difficult to express, unstable outside their native context, dependent on cofactors or partner proteins, or prone to disconnects between biochemical potency and cellular activity. In practice, the hardest part of an enzyme program is often not proving that the target is druggable, but finding differentiated chemical starting points quickly enough and validating them rigorously enough to justify downstream medicinal chemistry [4,5,9].
That is where DNA-encoded library screening has become especially compelling. DNA-encoded libraries (DELs) enable the pooled screening of extraordinarily large numbers of compounds through affinity selection, opening far broader chemical space than conventional one-compound-per-well formats. For enzyme targets, this breadth is highly valuable. DEL screening for enzymes can help identify new chemotypes for enzyme active site targeting, reveal allosteric binders, and support covalent enzyme inhibitor discovery strategies; all while consuming relatively little target material and allowing a binder-first route to enzyme hit discovery using DNA encoded libraries [10-14].
For organizations evaluating outsourcing enzyme inhibitor discovery, the practical appeal is straightforward: DEL can accelerate early enzyme drug discovery services by generating tractable hit matter before a full medicinal chemistry campaign is built around a target. And when paired with rapid off-DNA resynthesis, orthogonal enzymology, and cellular target-engagement assays, DEL screening for enzyme targets can become a highly efficient engine for discovering decision-ready starting points rather than just large volumes of screening data [10-18].
Why enzymes are highly druggable targets
The idea of “druggability” is often reduced to the presence of a pocket, but enzymes illustrate why the concept is broader than that. A druggable target must bind a small molecule with sufficient affinity and selectivity to produce a useful biological effect, yet enzymes frequently offer several ways to achieve that outcome at once [1]. Their active sites are shaped to recognize specific substrates or cofactors. Their catalytic residues create concentrated interaction points. Their conformational states can expose transient grooves or induce-fit pockets. And their function is often tightly connected to ligand binding, so occupancy can be translated into a measurable phenotypic effect more directly than for many structural or scaffolding proteins [1,6].
The record of approved drugs reinforces this. A major share of known molecular drug targets belong to enzyme families or enzyme-related mechanisms [2]. Kinases, proteases, polymerases, topoisomerases, phosphodiesterases, deacetylases, methyltransferases, and metabolic enzymes have all produced clinically important therapies. That success reflects both biological leverage and tractable chemistry. In other words, enzymes are not just abundant targets; they are targets for which medicinal chemistry has repeatedly demonstrated translational power [2-5].
Kinases are perhaps the clearest example. Protein phosphorylation controls growth, survival, immune signaling, DNA damage responses, metabolism, and cell fate. Unsurprisingly, abnormal kinase activity is implicated in cancer, inflammatory disease, fibrosis, and immune disorders. As a result, kinase inhibitor discovery has produced a deep and sophisticated knowledge base spanning ATP-competitive inhibitors, covalent binders, allosteric ligands, and conformationally selective chemotypes [3]. Proteases provide another archetype. They regulate clotting, apoptosis, extracellular remodeling, antigen processing, protein maturation, and pathogen replication. Protease inhibitor screening has yielded some of the field’s most powerful examples of mechanism-based drug discovery, but also some of its clearest lessons on the need for selectivity and context [4,5].
Importantly, the druggability of enzymes is not limited to orthosteric inhibition. Many enzymes are regulated allosterically, through distal pockets, protein-protein interfaces, lipid interactions, or conformational equilibria that shift catalytic competence. These mechanisms create additional opportunities when direct active-site competition is suboptimal. Allosteric modulation can improve subtype selectivity, avoid direct competition with high intracellular substrate concentrations, and provide access to differentiated pharmacology that is hard to achieve with classic active-site inhibitors [6]. For crowded families such as kinases and proteases, this can be commercially and scientifically decisive.
Covalent inhibition broadens the opportunity further. The modern resurgence of covalent drugs reflects a much more disciplined approach than earlier generations of reactive small molecules. Rather than relying on nonspecific reactivity, contemporary covalent inhibitor discovery uses tuned electrophiles, carefully positioned warheads, and structure-informed residue selection. For enzyme targets with catalytic serines, cysteines, or other suitably nucleophilic residues, covalent mechanisms can improve residence time, potency, and durability of target suppression [7,8]. This is particularly relevant to enzyme programs because catalysis often depends on exactly the kind of residue geometry that covalent chemistry can exploit.
Taken together, these features explain why enzymes remain such attractive drug targets. They are biologically central, chemically tractable, and mechanistically rich. But that same mechanistic richness also means discovery strategies need to be chosen carefully.
Key enzyme classes in small-molecule drug discovery
Although “enzyme” is a broad category, several target classes dominate practical small-molecule discovery.
Kinases remain one of the most important. The ATP-binding pocket offers a clear foothold for small molecules, but selectivity across the kinome is often difficult because the catalytic machinery is conserved. That challenge has pushed the field toward more nuanced strategies, including type II kinase inhibitors, allosteric binders, and covalent kinase inhibitors directed toward nonconserved residues [3,7,8]. For DEL screening for kinase targets, this means the value is not merely in identifying any ATP-site binder, but in surfacing chemotypes that offer credible selectivity and optimization headroom.
Proteases are similarly important but pose different challenges. Their active sites can be highly ligandable, yet peptide-mimetic chemistry can lead to poor permeability, off-target protease inhibition, or metabolic liabilities. Some protease families also rely on substrate-recognition motifs that are broad enough to create selectivity problems across related enzymes [4,5]. DEL screening for protease targets can therefore be especially valuable when it identifies nonclassical chemotypes, non-peptidic scaffolds, or covalent warhead placements that would be difficult to intuit from standard substrate-based design.
Beyond kinases and proteases, metabolic enzymes, epigenetic enzymes, deubiquitinases, helicases, polymerases, phosphatases, and oxidoreductases are all relevant enzyme classes for early discovery. In many of these families, the same central questions recur: is the best path an active-site inhibitor, an allosteric modulator, or a covalent mechanism? Is purified protein a good proxy for the relevant cellular state? Is selectivity the main technical hurdle, or is it cell penetration and target engagement? These are the kinds of questions that should shape how DEL screening for enzymes is designed from the outset.
What makes enzyme inhibitor discovery difficult
The phrase “enzymes are highly druggable” is true, but it can be misleading if it suggests low risk. In reality, enzyme inhibitor discovery fails for recurring reasons.
The first is selectivity. Many enzymes belong to large homologous families, and their active sites are built around conserved catalytic machinery. Kinases all bind ATP. Serine proteases share common catalytic logic. Metalloproteases often organize ligands around related metal-dependent motifs. A compound that looks excellent in a single biochemical assay can therefore prove unusable when challenged across close homologues, anti-targets, or broader selectivity panels [3-5]. This is especially relevant for commercial enzyme inhibitor discovery services, because early false confidence around selectivity can waste months of medicinal chemistry.
The second is the gap between biochemical inhibition and cellular activity. A molecule can inhibit purified enzyme strongly yet fail in cells because it is poorly permeable, rapidly effluxed, unstable, sequestered, or unable to compete with native substrate concentrations. Conversely, a compound with only modest biochemical potency can perform better than expected if it reaches the target efficiently or maintains long residence time [9,17,18]. For enzyme programs, the question is never just “does it inhibit?” but “does it engage the biologically relevant target state in a useful context?”
Residence time complicates the picture further. Classical IC50 or Kd values are informative, but they do not always capture how long a ligand remains bound under dynamic biological conditions. For enzymes operating in fluctuating substrate pools or signaling cascades, slower off-rates can improve pharmacological durability and, in some cases, functional selectivity [9]. That means early hit discovery should ideally generate multiple mechanistic hypotheses rather than optimizing around a single potency metric too early.
Assayability is another major issue. Some enzymes are straightforward to evaluate functionally. Others require unstable substrates, unusual cofactors, multi-component assemblies, membranes, or specific post-translational states. Some are better understood through conformational or binding readouts than through simple turnover assays. In those settings, a classical high-throughput functional screen may be expensive, artifact-prone, or simply too narrow in the chemistry it explores. DEL screening offers a useful alternative because it can begin with binder discovery and then move into biochemical and cellular confirmation once enriched chemotypes are in hand [10-16].
These realities are why enzyme hit discovery using DNA encoded libraries has become attractive. The value is not only scale; it is flexibility in how discovery is staged.
Why DEL screening is well suited to enzyme targets
DNA-encoded libraries consist of small molecules linked to DNA tags that encode their synthetic identity. Because each compound carries its own barcode, very large libraries can be pooled, screened together, and decoded by sequencing after selection. This architecture fundamentally changes the front end of hit discovery. Rather than testing thousands or tens of thousands of individually arrayed molecules, DEL enables the exploration of millions to billions of compounds in compact, pooled workflows [10-13].
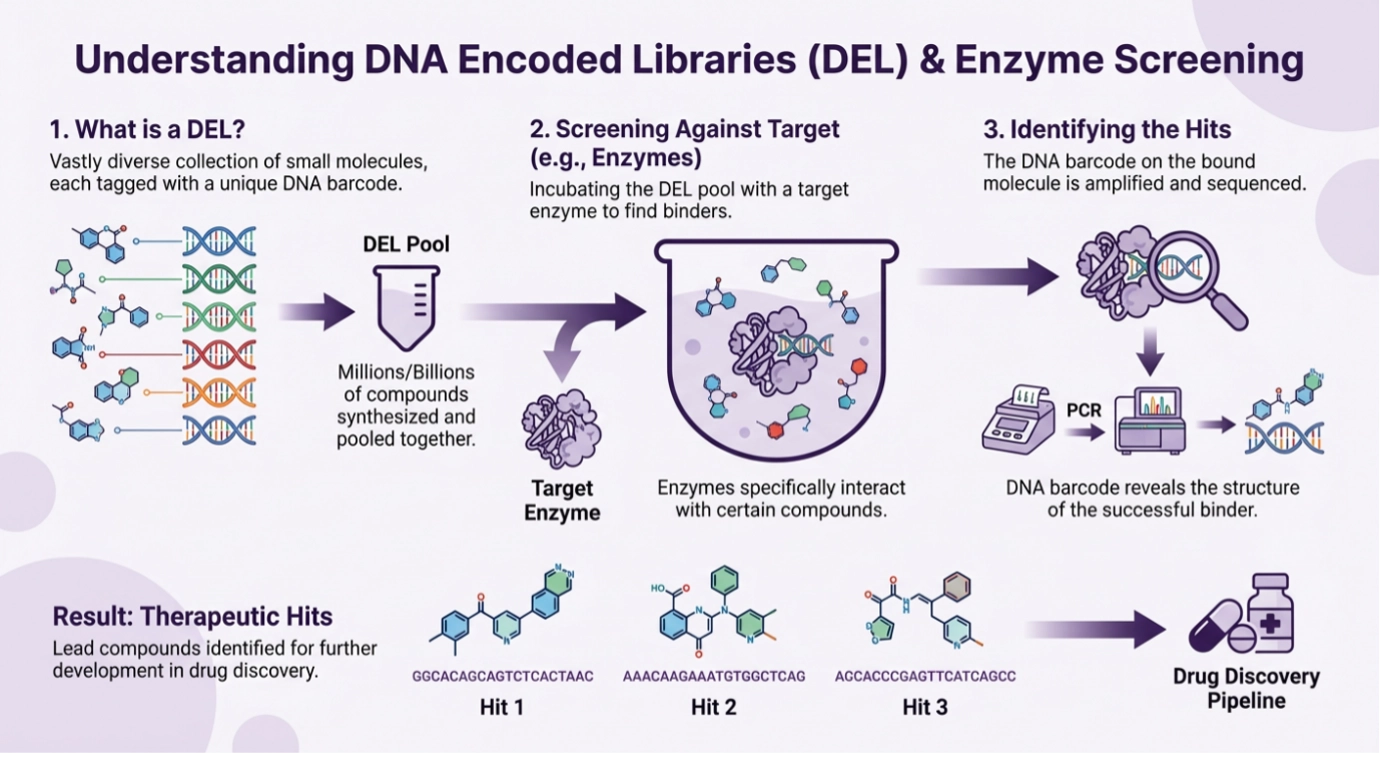
For enzymes, that scale matters because it makes it easier to search for rare but informative chemotypes. A modest screening deck may be enough to find a familiar active-site inhibitor for a well-studied kinase. It is much less likely to reveal new binding modes, differentiated selectivity escape routes, or unusual allosteric series. DEL breadth increases the odds of finding those less obvious opportunities, which is one reason DNA encoded library screening services are increasingly relevant to early-stage enzyme programs [10-14].
Another strength is that DEL is, at its core, a binding-based technology. For enzyme drug discovery, this is often an advantage rather than a limitation. Binding can be discovered before the perfect functional assay exists, before the best substrate pair is optimized, or before the team decides whether orthosteric, allosteric, or covalent inhibition is the right objective. In that sense, DEL screening for enzymes is often best understood as a front-end chemical space exploration tool that feeds a richer validation cascade [10-16].
This is particularly powerful for enzyme active site targeting. Enzyme pockets often reward modular optimization around a recognition core, and DEL library design can exploit that logic by combining privileged motifs with diverse appendages or warheads. The field’s early landmark studies showed that DEL could yield genuine enzyme-active chemotypes. Clark and colleagues demonstrated that DNA-encoded small-molecule libraries could identify inhibitors of Aurora A kinase and p38 MAP kinase, helping establish DEL as a practical route to enzyme hit discovery rather than just a theoretical screening format [15].
Since then, DEL has advanced far beyond generic affinity selections. Reviews collectively show how DNA-encoded library technology has matured in chemistry, selection design, and post-selection strategy [10-14]. These advances matter directly for enzymes because not every enzyme target behaves well in the same selection setup. Some are soluble and stable. Others require more nuanced formats or more careful off-DNA follow-up. The most productive DEL campaigns are therefore not generic – they are deliberately matched to the biology of the target.
Off-DNA strategies also strengthen the platform. Hackler and colleagues showed how off-DNA DNA-encoded library affinity screening can help interrogate compounds in a context that is closer to classical assay formats, reducing ambiguity around the effect of the DNA tag itself [16]. For enzyme programs, that can be especially useful when transitioning from enrichment data to genuine inhibitor confirmation.
In-cell DEL and physiologically relevant enzyme hit discovery
One reason early enzyme programs stall is that the recombinant protein used in screening is not a faithful representation of the biologically relevant target state. The active conformation may depend on a cofactor, membrane association, binding partner, cellular redox state, or post-translational modification that is difficult to preserve in vitro. Even when purified protein is available, biochemical activity does not guarantee that a hit will engage the target in cells [17,18].
This is where more physiologically relevant DEL strategies become interesting. In-cell DEL concepts aim to move hit finding closer to the environment where the target operates [25]. More broadly, cell-relevant follow-up assays such as CETSA can test whether a compound physically engages its target in cells rather than only inhibiting a purified enzyme in a tube [17,18]. Martinez Molina and colleagues first showed that the cellular thermal shift assay can monitor drug-target engagement in cells and tissues, and Jafari and colleagues later described the method in practical detail [17,18]. For enzyme inhibitor discovery, that matters because it helps bridge one of the biggest early questions: not just “does the compound bind?” but “does it bind under intracellular conditions that matter?”
Scientifically, the combination of DEL screening and early cellular target engagement is compelling. DEL can search broad chemical space quickly, then off-DNA compounds can be triaged using biochemical inhibition, orthogonal binding assays, and cell-based target engagement or pathway readouts. That workflow is particularly attractive for challenging enzymes, for membrane-associated proteins, and for targets where purified protein is not the most informative representation of the druggable state.
For a company like Vipergen, which emphasizes both Binder Trap Enrichment® and Cellular Binder Trap Enrichment® in its platform positioning, this creates a strong content angle: DEL screening does not need to stop at purified proteins, and physiologically relevant screening contexts can help prioritize enzyme binders that have a better chance of surviving downstream attrition [23-25].
From DEL binders to validated enzyme inhibitors
After a DEL screen, enriched compounds or chemotypes need to be resynthesized off-DNA and tested in orthogonal assays. The first question is whether the compound truly binds the target without the DNA tag. The second is whether that binding translates into meaningful modulation of enzyme function. The third is whether the mechanism is orthosteric, allosteric, or covalent. And the fourth is whether the compound has early signs of tractability in terms of selectivity, cellular engagement, and chemistry follow-up [10-18].
This sequence matters because enzyme binders are not automatically useful inhibitors. A compound may bind in the active site without blocking catalysis under physiological substrate conditions. It may bind a nonproductive conformation. It may show good enzymology but poor cellular exposure. Or it may be a very valuable allosteric ligand that would be missed if the wrong functional assay is used too early. The practical goal in early enzyme drug discovery is therefore not only to confirm activity, but to clarify mechanism and optimization potential.
That is why the most credible enzyme inhibitor discovery services usually combine DEL screening with rapid off-DNA resynthesis and focused medicinal chemistry follow-up. Once series are confirmed, biochemical potency, counterscreens, anti-target profiling, and early cellular assays can be layered into separate attractive starting points. In a commercial setting, the quality of this transition from sequencing output to decision-ready compounds often matters more than the raw number of enriched hits.
DEL screening for kinase and protease targets
Kinases and proteases make especially useful case studies because they represent two high-value enzyme classes with distinct discovery constraints.
For kinases, DEL screening can help in at least three ways. First, it can identify fresh ATP-site chemotypes in cases where known hinge-binding space is crowded. Second, it can support covalent kinase inhibitor discovery when the target offers a suitably positioned residue. Third, it can help surface chemotypes that are worth testing against inactive or allosteric conformations rather than only the canonical active state [3,7,8,15,20]. The early DEL work by Clark et al. on Aurora A and p38 established that kinase-focused DEL screening can produce real enzyme-active matter [15], while later triazine-based covalent DEL work by Li et al. extended the concept into covalent discovery against enzymes such as BTK and JAK3 [20].
Proteases illustrate a somewhat different challenge. Their active sites are often highly tractable, but selectivity and developability can be difficult. Classical peptide-like protease inhibitors may bind well but perform poorly in broader drug-like optimization. This makes protease inhibitor screening a good fit for focused DEL design, where substrate-inspired recognition can be balanced against the need for differentiated, non-peptidic, or covalent chemistry. Dawadi and colleagues showed that a protease-focused DNA-encoded chemical library could yield potent thrombin inhibitors, demonstrating the value of class-biased library design for enzyme discovery [19]. More recently, DEL campaigns against SARS-CoV-2 main protease identified both covalent and non-covalent inhibitor series, reinforcing that even within a single protease target, multiple DEL-enabled discovery routes can be productive [21,22].
DEL screening for enzymes works best when it is not generic. The ideal library, selection conditions, and follow-up assays depend on the enzyme class, the mechanism sought, and the practical bottleneck in the program. For DEL screening for kinase targets, selectivity and differentiation may dominate. For protease inhibitor discovery, the emphasis may be on nonclassical chemistry, covalent logic, or avoiding peptide liabilities. In both cases, the strength of DEL lies in giving the program more chemical options at the point where good options matter most.
What to look for when outsourcing enzyme inhibitor discovery
For teams considering outsourcing enzyme inhibitor discovery, the right partner is not simply the one with the biggest library. The better question is whether the provider can convert enzyme hit discovery into validated and actionable chemistry.
That usually means five things. First, the partner should understand the biology well enough to choose the correct screening hypothesis: active-site inhibitor, allosteric binder, covalent mechanism, or a staged combination of these. Second, the provider should have a fast path from DEL enrichment to off-DNA confirmation. Third, library design should be connected to the target class rather than purely generic. Fourth, selectivity should be considered early, especially for conserved enzyme families. And fifth, the service should not stop at sequencing output; it should support the progression from hit identification to early lead-quality matter [4-8,10-18].
Vipergen’s high-fidelity DEL technologies, screening in living cells via cBTE,[25] and medicinal chemistry support for off-DNA validation and early series progression [23,24]. Furthermore the cBTE technology allows for multiplex screening against counter targets, which means that selectivity can be dialed into the screening.
Conclusion
Enzymes remain among the most valuable targets in drug discovery because they combine biological centrality with multiple chemically tractable modes of control. Active-site inhibition, allosteric modulation, and covalent targeting all remain highly relevant strategies across kinases, proteases, metabolic enzymes, and many other enzyme classes [1-8].
But the practical challenge in enzyme programs is rarely whether chemistry can bind the target at all. The challenge is finding differentiated series quickly, confirming mechanism rigorously, and prioritizing molecules that stand a realistic chance of working in cells. DNA encoded library screening is especially well suited to that front end of discovery because it enables broad chemical-space exploration while preserving a clear path into biochemical, biophysical, and cellular validation [10-18].
For that reason, DEL screening for enzymes is becoming an increasingly attractive route for enzyme hit discovery using DNA encoded libraries, whether the program is focused on kinase inhibitor discovery, protease inhibitor screening, enzyme active site targeting, or covalent enzyme inhibitors DEL strategies [15,19-22]. And for companies evaluating enzyme inhibitor discovery services, the most valuable DEL partner will be the one that combines screening scale with target-class understanding, rapid off-DNA follow-up, and a practical path from hit to validated inhibitor [23,24].
References