



Protein-Protein Interactions as Drug Targets: How DNA-Encoded Library Screening Opens New Paths for “Undruggable” PPIs

Protein-protein interactions (PPIs) sit at the center of cellular biology. They assemble signaling complexes, control gene expression, regulate protein stability, and coordinate nearly every process that keeps cells alive. When those interactions become dysregulated (e.g. through mutation, overexpression, mislocalization, or viral hijacking) they can drive disease. That is why PPIs have long been recognized as an enormous therapeutic opportunity. Yet, for decades, many of the most compelling PPIs were labeled “undruggable.” The underlying reason is structural: a large fraction of PPIs relies on broad, relatively flat contact surfaces rather than deep pockets that small molecules easily occupy. Reviews frames both the challenge and the opportunity: PPI interfaces can be difficult, but they often contain “hotspots” and inducible pockets that can be exploited if you have the right discovery strategy. [1–3]
This is where modern hit-finding approaches matter and why DNA-encoded libraries (DELs) have become increasingly relevant for protein-protein interaction inhibitors and small molecule PPI inhibitors discovery. DELs enable affinity-based screening of extraordinarily large chemical collections, helping identify chemotypes that can engage challenging protein interaction binding sites, including PPI hotspots and allosteric pockets. [12–14]
For organizations working on undruggable targets drug discovery, the question is no longer “Are PPIs druggable?”. It is “Which PPI modality fits the biology, and what screening format best captures that biology?” Increasingly, that includes DEL screening for protein-protein interactions, and in some cases, intracellular or cell-context selections that preserve native conformations, post-translational modifications, cofactors, and competing binding partners. [15–17]
This article explains:
- why PPIs matter biologically and therapeutically,
- what makes PPI interfaces difficult (and what makes them tractable),
- how allosteric PPI modulators and hotspot binders expand the solution space,
- why DEL can be particularly effective on large, shallow interfaces, and
- how an intracellular/cellular DEL workflow can support hit discovery for challenging PPI targets and downstream optimization.
PPIs in Cellular Biology: Why They Make Powerful (and Difficult) Drug Targets
PPIs are not “special cases” – they are the default mechanism by which cells build functional machines. Enzymes often act in complexes; transcription factors recruit co-activators and repressors; kinases dock to scaffolds; ubiquitin ligases recognize substrates via protein interfaces. The clinical relevance is straightforward: if a disease state depends on a pathologic interaction, modulating that interaction can be as direct and causal as inhibiting an enzyme active site.
Dynamic and context-dependent interactions
Many PPIs are transient, forming only in particular signaling states, in specific subcellular compartments, or at defined times in the cell cycle. Others are stable and obligate (e.g., structural complexes). This matters because a screening campaign against a purified fragment can miss (or invent) binding opportunities that do not exist in the cellular state. Reviews of PPI drug discovery emphasize that “what you screen” (constructs, conformations, binding partners) can be as important as “what you screen with.” [1–3]
PPIs in disease mechanisms
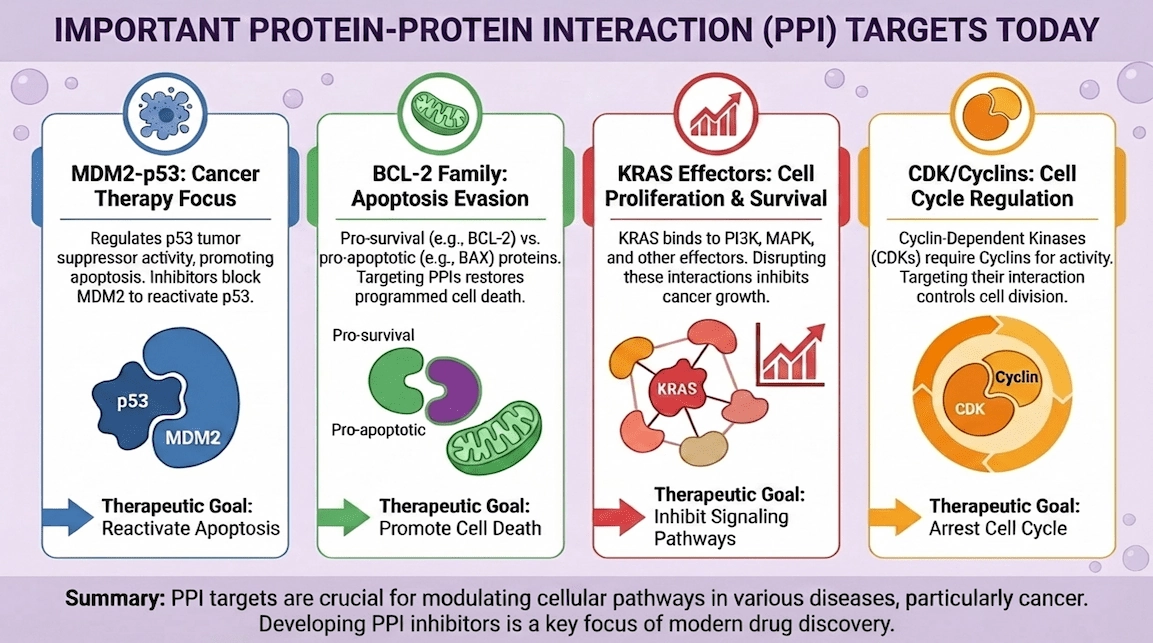
Cancer provides familiar examples: the p53 tumor suppressor is restrained by the MDM2 protein through a well-defined interaction; small-molecule antagonists of that interaction validated the concept that even high-value PPIs can be addressed with drug-like ligands. [8]
Apoptosis is another canonical area: BCL-2 family proteins control survival by binding BH3 helices; the BCL-2 inhibitor venetoclax (ABT-199) shows how a PPI-like groove can become a tractable small-molecule target with sufficient structural understanding and chemistry. [9]
What Makes PPIs “Undruggable” and What Makes Them Tractable
The phrase “undruggable targets drug discovery” persists because many PPIs do not present the classical features of enzyme active sites. Three structural themes show up repeatedly.
1) Large and adaptive interfaces
Many PPIs bury ~1,500–3,000 Ų of surface area, often spread across discontinuous residues. That looks mismatched to a 300–500 Da molecule. However, the key insight from hotspot research is that binding energy is not evenly distributed. A minority of residues can contribute most of the interaction free energy. [4]
2) Hotspots and “protein interaction binding sites”
Hotspots are clusters of residues where a small chemical footprint can have outsized functional impact. The classic hotspot paper showed that alanine-scanning often reveals a small set of energetically dominant positions. [4]
For drug discovery, this means a large “flat” interface may still contain localized pockets or crevices where a ligand can anchor, especially if the ligand can mimic key hydrophobic/aromatic hotspot contributions.
3) Cryptic pockets and conformational selection
Some PPIs appear pocketless until a ligand (or protein partner) induces or stabilizes a conformation that reveals a pocket. These are often called cryptic binding sites. Reviews and systematic mapping efforts highlight that cryptic pockets can be widespread and can create new entry points for PPI modulation especially for allosteric approaches. [5,6]
Practical implication: many “undruggable” PPIs are not truly undruggable – they are undersampled. Success is often about screening strategies that can find rare chemotypes that bind shallow hotspots, stabilize transient pockets, or engage allosteric sites.
Classes of PPIs That Are Especially Suitable for Small-Molecule Modulation
Not all PPIs are equal from a ligandability perspective. If your goal is small molecule PPI inhibitors discovery, it helps to recognize which interaction archetype you are dealing with.
Transient vs. stable interactions
- Transient PPIs (common in signaling) often rely on short linear motifs, phosphorylation-dependent docking, or weak interactions that become strong in complexes. These can be susceptible to inhibitors that block recruitment or disrupt assembly.
- Stable/obligate complexes may be harder to disrupt directly, but can still be modulated allosterically, destabilized indirectly, or addressed via molecular glue/degrader strategies. [1–3]
Groove-and-helix interfaces
A frequent “druggable PPI” pattern is an amphipathic helix docking into a groove (e.g., BCL-2 family, MDM2/p53). These are often more pocket-like and have produced multiple clinical-stage ligands. [7–9]
Hotspot-dominated flat interfaces
These are the canonical “hard” PPIs: shallow, extended surfaces where no single deep pocket is obvious. Here, discovery frequently relies on:
- finding hotspot anchors (often aromatic/hydrophobic),
- growing or linking fragments, or
- identifying allosteric sites that indirectly modulate the interaction. [1–6]
Allosteric and indirect modulation
Allosteric modulation can be a powerful way to avoid the flattest part of the interface. A ligand binds elsewhere, shifts conformational equilibria, and changes the interaction. This is especially attractive when the orthosteric interface lacks pockets or when selectivity is challenging. The PPI field has also matured beyond inhibitors alone: stabilization, induced proximity, and “molecular glue” mechanisms are increasingly recognized as viable therapeutic modes. [7,10,11]
Modalities: Inhibitors, Stabilizers, and Molecular Glues
When people search for protein-protein interaction inhibitors, they often mean “blockers.” But many high-value PPI programs now consider multiple modalities, because the biology can favor one mechanism over another.
Orthosteric inhibitors (direct disruption)
These bind at the interface and compete with the partner protein. MDM2/p53 antagonists are a flagship example, demonstrating that a small molecule can occupy a key interface pocket and reactivate tumor suppressor signaling. [8]
Allosteric PPI modulators
These bind outside the interface and reduce (or sometimes enhance) binding indirectly. This can be advantageous when:
- the interface is too flat,
- selectivity is difficult at the orthosteric site, or
- you need partial modulation rather than full shutdown. [7]
Stabilizers and “molecular glues”
Stabilization is an underappreciated concept: instead of disrupting a PPI, you strengthen a beneficial interaction or lock a complex in a nonproductive state. A well-known theme in chemical biology is that small molecules can stabilize interactions as part of their mechanism. Reviews highlight this as a distinct and valuable design space. [10]
“Molecular glues” are a related idea: a ligand induces or stabilizes a new protein-protein association. The lenalidomide/cereblon story illustrates a glue-like mechanism where a small molecule alters an E3 ligase’s substrate recognition to drive selective degradation of transcription factors, hereby demonstrating how modulating protein interfaces can rewire cell biology. [11]
Why this matters for screening: a discovery platform that can detect binders to hotspots and allosteric pockets (and that can be run in formats close to physiology) expands your odds of finding workable starting points for challenging PPIs.
Why DNA-Encoded Libraries Are a Natural Fit for PPI Hit Discovery
DEL’s core advantage: scale with affinity-based readout
The original concept of encoding small molecules with amplifiable DNA information emerged in the early 1990s, linking combinatorial chemistry with genetic-style selection. [12]
Modern DEL platforms routinely enable screening of extremely large chemical spaces, with the practical readout being enrichment of DNA barcodes after binding/selection. Reviews in Nature Reviews Drug Discovery and ACS Pharmacology & Translational Science summarize the evolution of DEL chemistry, selection formats, and downstream translation to off-DNA molecules. [13,14]
For PPIs, scale matters because:
- true hotspot binders can be rare,
- shallow interfaces often reward “lucky” shape complementarity, and
- allosteric pockets can be subtle and hard to predict without empirical screening.
DEL as a solution for large, shallow interfaces
Large, shallow interfaces challenge classical HTS because typical screening collections were historically biased toward enzyme-like pockets. DELs can be designed with PPI-relevant features for example:
- higher 3D character (sp³-rich scaffolds),
- macrocyclic or constrained motifs,
- aromatic-rich hotspot mimics,
- electrophile-free chemotypes for broad compatibility (or purpose-built covalent strategies where appropriate),
- fragment-like elements that can be elaborated.
While DEL hits still require careful validation, the platform is particularly attractive for hit discovery for challenging PPI targets because it increases the odds of sampling unusual geometries that match hotspot landscapes.
Hotspot binding + allostery: two routes to PPI control
One of the strongest conceptual matches between DEL and PPIs is that DEL can yield:
- direct interface binders (hotspot blockers), and
- allosteric binders that reshape the interface or partner affinity.
Cryptic pockets and conformational selection provide a structural explanation for why allosteric DEL hits can appear even when an interface looks “flat” in a single structure. [5,6]
Intracellular and Cell-Context DEL Screening: Why “Where You Screen” Matters for PPIs
Vipergen’s focus on DEL screening against essentially any protein target naturally includes PPIs. For many intracellular PPIs, the biologically relevant state depends on:
- native folding and quality-control pathways,
- post-translational modifications (phosphorylation, acetylation, ubiquitination),
- compartmental localization,
- endogenous binding partners that compete or cooperate.
For these reasons, cell-context screening formats can be compelling complements to purified-protein selections.
Evidence that DEL selections can be done in cellular settings
Peer-reviewed work has demonstrated multiple strategies for bringing DEL selection closer to cellular biology:
- Cell-based DEL selections: early demonstrations showed that DEL selections can be performed in cell contexts rather than only on purified proteins. [15]
- DEL screening inside living cells: subsequent work advanced the concept further, showing selection/screening formats compatible with intracellular environments. [16]
- Live-cell targeting of endogenous proteins: approaches have also been reported for selections against targets on or in living cells (including endogenous membrane proteins), supporting the broader theme that DEL can move beyond immobilized purified proteins. [17]
These papers do not “solve” every intracellular PPI problem – delivery, nonspecific binding, and assay design remain nontrivial – but they establish an important point: DEL screening for protein-protein interactions can be designed in formats that preserve more native biology than classical in vitro-only workflows.
Why intracellular DEL can be especially valuable for PPIs
For PPIs, the binding surface can be:
- transiently formed only in a signaling state,
- reshaped by phosphorylation or ligand binding,
- occluded unless a complex assembles,
- altered by crowding, local concentration, or scaffolding proteins.
Cell-context selection can help capture:
- the correct complex (or complex equilibrium),
- realistic conformational ensembles,
- functional coupling to downstream pathways (if the selection is designed around functional states).
Practical PPI Target Classes Well-Suited to Cellular or Intracellular DEL Approaches
Not every PPI program needs in-cell selection. But for certain classes, the rationale is strong:
- Complexes that require cofactors or PTMs
If the interface forms only after phosphorylation (or another modification), screening a static purified construct can be misleading. - Targets with conformation-sensitive pockets
Cryptic pockets may appear only in specific cellular states. [5,6] - Competitive interaction networks
Many PPIs are part of hubs. In-cell formats can bias toward ligands that function in the presence of endogenous competitors, often a major gap between biochemical binding and cellular efficacy. - Allosteric modulation as primary strategy
If you already believe the best route is an allosteric site, preserving native conformational equilibria can help surface those binders.
A DEL Workflow for PPI Programs (Designed to Support Both Commercial and Informational Needs)
DNA encoded library PPI screening can be integrated into a modern PPI program – starting from hit finding and moving toward validated chemical matter.
1) Define the PPI hypothesis and intervention strategy
Before screening, clarify what “modulation” means for the biology:
- Do you need an inhibitor (disruptor), stabilizer, or molecular glue-like effect?
- Is orthosteric blocking plausible, or is an allosteric strategy more realistic?
- What functional readout will matter downstream?
This framing reduces wasted cycles and helps interpret DEL enrichments: a binder might not be an inhibitor but could still be a valuable allosteric PPI modulator or a handle for induced proximity strategies. [7,10,11]
2) Choose the selection format: purified complex, reconstituted system, or cellular context
Options often include:
- purified partner proteins (orthosteric hotspot hunting),
- pre-assembled complexes (to bias toward interface pockets present only in the complex),
- competitive selections (add a peptide/protein competitor to enrich higher-affinity or orthosteric binders),
- cellular/intracellular selection formats (to preserve native biology). [15–17]
A useful mental model is the harder the PPI and the more state-dependent the interface, the more valuable a native-like format becomes.
3) Library strategy: match chemical space to PPI reality
For PPIs, “bigger library” is helpful, but “better-shaped library” is often decisive. DEL design considerations commonly include:
- 3D shape and rigidity: helps engage shallow sites and hotspots.
- Aromatic/hydrophobic anchors: frequently match hotspot residue chemistry. [4]
- Macrocycles/constrained motifs: can cover more surface area with fewer rotatable bonds.
- Exit vectors for SAR: ensure hits can be resynthesized and optimized off-DNA.
This is an important bridge between informational and commercial intent: companies searching for PPI drug discovery services are often specifically looking for providers who understand how to tailor libraries and selection conditions for PPI-type binding sites rather than enzyme pockets.
4) Selection execution and decoding
In a typical DEL campaign:
- the library is incubated with the target (or complex),
- non-binders are washed away (with conditions tuned to preserve meaningful interactions),
- binders are recovered,
- DNA tags are PCR amplified and sequenced to identify enriched structures. [13,14]
For PPIs, selection design often includes extra controls:
- counter-selections to remove sticky scaffolds,
- partner-only or target-only controls to map binding specificity,
- competition with peptide motifs to distinguish orthosteric binders.
5) Hit triage: from enriched barcode to real chemistry
DEL data are powerful, but they are not “the answer” on their own. Key steps include:
- resynthesis of top hits off-DNA,
- orthogonal binding assays (SPR, BLI, ITC, NMR, DSF – depending on target),
- functional assays that read out PPI modulation (biochemical and cellular).
This is the critical bridge to hit discovery for challenging PPI targets: what matters is not just binding, but a validated mechanism that can be optimized.
6) Mechanism mapping: orthosteric vs allosteric vs stabilization
For PPIs, you typically want to know:
- Does the ligand compete with the partner?
- Does it bind elsewhere and modulate affinity?
- Does it stabilize a particular complex state?
Examples across the literature highlight that each mechanism can be therapeutically useful. [7,10,11]
7) Medicinal chemistry and expansion
Once a hit series is validated:
- explore SAR around hotspot anchors or allosteric pockets,
- improve potency and selectivity,
- optimize cell permeability (particularly important for intracellular PPIs),
- maintain ligand efficiency while expanding interface coverage.
How AI-Enabled Narratives Fit DEL + PPI Programs
Many teams want “AI-enabled discovery” not as branding, but as practical leverage:
- prioritize PPIs by tractability signals,
- predict interface structures and possible pockets,
- interpret DEL enrichment patterns at scale.
A concrete example of enabling infrastructure is AlphaFold-level structure prediction, which dramatically expanded access to structural hypotheses for proteins lacking experimental structures. While AlphaFold does not automatically solve PPI pocket discovery, it can accelerate model generation, interface mapping, and hypothesis-driven selection design – especially when paired with experimental validation. [18]
In DEL + PPI workflows, AI/ML is often most valuable in:
- target triage: which PPIs have plausible ligandable hotspots or allosteric pockets?
- selection design: which constructs and conformations should be prioritized?
- data mining: identifying meaningful clusters in DEL enrichments and filtering artifacts.
- SAR acceleration: learning from early hit series to propose analogs for follow-up synthesis.
Frequently Asked Questions
Many are. The field shifted from “impossible” to “selectively tractable” as hotspot biology and successful programs accumulated. [1–4]
- Inhibitors typically block the interface directly (orthosteric competition).
- Allosteric modulators bind elsewhere and change the interaction indirectly (often by shifting conformations or stabilizing/destabilizing states). [7]
DEL gives you access to far larger chemical diversity with affinity-based selections, which is valuable when true binders to shallow sites are rare. DEL reviews describe how scale and selection formats have matured, including movement toward more biologically relevant contexts. [13–17]
Peer-reviewed studies support selection/screening formats that operate in cell contexts, including intracellular environments and live-cell targeting strategies. [15–17]
For challenging PPIs, look for teams that can:
- design selection formats around complexes and competition,
- interpret PPI-relevant binding patterns (hotspots, shallow sites, allostery),
- validate hits with orthogonal assays and functional readouts,
- support medicinal chemistry expansion toward cell-active matter.
Takeaway: DEL Expands the Search Space for PPI Drug Discovery
The modern view of PPIs is pragmatic: many are difficult, but difficulty is not a dead end. Hotspots concentrate binding energy, cryptic pockets create hidden opportunities, and allosteric mechanisms offer alternate routes when the interface is too flat. [1–7]
For discovery teams pursuing undruggable targets drug discovery, DEL offers a compelling engine for small molecule PPI inhibitors discovery and for uncovering allosteric PPI modulators – especially when combined with screening formats designed to preserve relevant biology. Peer-reviewed work now supports cellular and intracellular DEL paradigms, strengthening the case for DNA encoded library PPI screening as a practical route to hit discovery for challenging PPI targets. [13–17]
References
- Wells J.A., McClendon C.L., Reaching for high-hanging fruit in drug discovery at protein-protein interfaces, Nature (2007), 450(7172), 1001–1009. DOI: 10.1038/nature06526. https://doi.org/10.1038/nature06526
- Arkin M.R., Wells J.A., Small-molecule inhibitors of protein–protein interactions: progressing towards the dream, Nat Rev Drug Discov (2004), 3(4), 301–317. DOI: 10.1038/nrd1343. https://doi.org/10.1038/nrd1343
- Scott D.E., Bayly A.R., Abell C., et al., Small molecules, big targets: drug discovery faces the protein-protein interaction challenge, Nat Rev Drug Discov (2016), 15(8), 533–550. DOI: 10.1038/nrd.2016.29. https://doi.org/10.1038/nrd.2016.29
- Bogan A.A., Thorn K.S., Anatomy of hot spots in protein interfaces, J Mol Biol (1998), 280(1), 1–9. DOI: 10.1006/jmbi.1998.1843. https://doi.org/10.1006/jmbi.1998.1843
- Vajda S., Beglov D., Wakefield A.E., et al., Cryptic binding sites on proteins: definition, detection, and druggability, Curr Opin Chem Biol (2018), 44, 1–8. DOI: 10.1016/j.cbpa.2018.05.003. https://doi.org/10.1016/j.cbpa.2018.05.003
- Cimermancic P., Weinkam P., Rettenmaier T.J., et al., CryptoSite: expanding the druggable proteome by characterization and prediction of cryptic binding sites, J Mol Biol (2016), 428(4 Pt A), 709–719. DOI: 10.1016/j.jmb.2016.01.029. https://doi.org/10.1016/j.jmb.2016.01.029
- Lu H., Zhou Q., He J., et al., Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials, Sig Transduct Target Ther (2020), 5, 213. DOI: 10.1038/s41392-020-00315-3. https://doi.org/10.1038/s41392-020-00315-3
- Vassilev L.T., Vu B.T., Graves B., et al., In vivo activation of the p53 pathway by small-molecule antagonists of MDM2, Science (2004), 303(5659), 844–848. DOI: 10.1126/science.1092472. https://doi.org/10.1126/science.1092472
- Souers A.J., Leverson J.D., Boghaert E.R., et al., ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets, Nat Med (2013), 19(2), 202–208. DOI: 10.1038/nm.3048. https://doi.org/10.1038/nm.3048
- Thiel P., Kaiser M., Ottmann C., Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery?, Angew Chem Int Ed Engl (2012), 51(9), 2012–2018. DOI: 10.1002/anie.201107616. https://doi.org/10.1002/anie.201107616
- Krönke J., Udeshi N.D., Narla A., et al., Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells, Science (2014), 343(6168), 301–305. DOI: 10.1126/science.1244851. https://doi.org/10.1126/science.1244851
- Brenner S., Lerner R.A., Encoded combinatorial chemistry, Proc Natl Acad Sci USA (1992), 89(12), 5381–5383. DOI: 10.1073/pnas.89.12.5381. https://doi.org/10.1073/pnas.89.12.5381
- Goodnow R.A. Jr., Dumelin C.E., Keefe A.D., DNA-encoded chemistry: enabling the deeper sampling of chemical space, Nat Rev Drug Discov (2017), 16(2), 131–147. DOI: 10.1038/nrd.2016.213. https://doi.org/10.1038/nrd.2016.213
- Gironda-Martínez A., Donckele E.J., Samain F., et al., DNA-Encoded Chemical Libraries: A Comprehensive Review with Succesful Stories and Future Challenges, ACS Pharmacol Transl Sci (2021), 4(4), 1265–1279. DOI: 10.1021/acsptsci.1c00118. https://doi.org/10.1021/acsptsci.1c00118
- Cai P., Tagore D.M., Chatterjee S., et al., Selection of DNA-Encoded Libraries to Protein Targets within and on Living Cells, J Am Chem Soc (2019), 141(43), 17057–17061. DOI: 10.1021/jacs.9b08085. https://doi.org/10.1021/jacs.9b08085
- Petersen L.K., Blakskjær P., Chaikuad A., et al., Screening of DNA-Encoded Small Molecule Libraries inside a Living Cell, J Am Chem Soc (2021), 143(7), 2751–2756. DOI: 10.1021/jacs.0c09213. https://doi.org/10.1021/jacs.0c09213
- Huang Y., Meng, L., Nie, Q., et al., Selection of DNA-encoded chemical libraries against endogenous membrane proteins on live cells, Nat Chem (2021), 13(1), 77–88. DOI: 10.1038/s41557-020-00605-x. https://doi.org/10.1038/s41557-020-00605-x
- Jumper J., Evans R., Pritzel A., et al., Highly accurate protein structure prediction with AlphaFold, Nature (2021), 596(7873), 583–589. DOI: 10.1038/s41586-021-03819-2. https://doi.org/10.1038/s41586-021-03819-2