Transporters as Drug Targets: Strategies for Modern Ligand Discovery
Transporters as Drug Targets: Strategies for Modern Ligand Discovery
Why transporters matter in drug discovery
Transporters have become an important focus in drug discovery, not just a side topic in ADME or drug-drug interaction work. That change reflects a basic point: transporters do more than influence where a drug goes in the body. Many of them are directly involved in disease biology and can be addressed with small molecules. By controlling the movement of nutrients, metabolites, ions, neurotransmitters, and xenobiotics across membranes, transporters affect cell survival, signaling, metabolism, and treatment response in a very direct way [1,2].
This has clear implications across several therapeutic areas. In cancer, transporters can determine whether a tumor cell has access to amino acids, glucose, or cystine. In the CNS, they control how long neurotransmitters remain available at the synapse. In kidney and metabolic disease, they influence glucose reabsorption and broader solute handling. In each case, changing transporter activity can alter disease biology itself rather than only changing drug exposure [1,2].
The scale of the opportunity is substantial. The human SLC superfamily contains more than 400 genes, many still poorly explored despite clear links to disease and pharmacology [1,2]. ABC transporters add another clinically important group, especially in efflux, tissue protection, and multidrug resistance [1]. For drug discovery teams, that means transporters represent a broad target class with room for new chemistry, new biology, and differentiated therapeutic strategies [1,2].
Transporters control access to key cellular inputs
Transporters occupy a different position in biology than receptors or enzymes. A receptor transmits information. An enzyme converts a substrate. A transporter determines what enters the cell, what leaves it, and what accumulates inside. That gives transporter pharmacology a particularly direct effect on cell state. In the right disease setting, even partial modulation can be enough to change growth, signaling, stress tolerance, or drug response [1].
SLC and ABC transporters are central target families
Two transporter families matter most in current drug discovery discussions.
- SLC transporters are involved in nutrient uptake, metabolite exchange, ion transport, and neurotransmitter handling [1,2].
- ABC transporters are best known for ATP-dependent efflux and for their role in drug resistance and tissue protection [1].
These families differ in mechanism and in the types of drugs they may support, but both include proteins with strong biological rationale and growing pharmacological interest [1,2].
Transporters are already validated drug targets
Transporters are not a speculative target class. There are already well-established clinical examples where transporter modulation leads to therapeutic benefit.
Clinical proof in CNS and cardiometabolic disease
One of the clearest CNS examples is the serotonin transporter, SERT. Common antidepressants work by blocking serotonin reuptake, increasing serotonin signaling in the synapse. Structural studies of human SERT have shown how antidepressants bind and stabilize defined conformational states, while also identifying an allosteric site that can affect ligand behavior [3]. That matters because it shows transporter pharmacology can involve more than simple substrate-pocket competition.
The case is even stronger in metabolic and renal disease. SGLT2 inhibitors changed the treatment of type 2 diabetes and later showed broader benefit in cardiorenal disease. In the clinical trial EMPA-REG OUTCOME, empagliflozin reduced cardiovascular events and mortality in patients with type 2 diabetes at high cardiovascular risk [4]. In CREDENCE, canagliflozin reduced the risk of kidney failure and cardiovascular events in patients with diabetic nephropathy [5]. These are strong examples of how transporter-directed pharmacology can lead to clinically important outcomes [4,5].
Growing relevance in oncology
Oncology is one of the most active areas for transporter drug discovery. Tumor cells often depend on transporters to maintain nutrient supply, redox balance, and resistance to stress. LAT1, also called SLC7A5, is a good example. It transports large neutral amino acids and supports nutrient-dependent growth pathways in many tumors. A phase I study of JPH203 showed that a selective LAT1 inhibitor could be advanced into the clinic for patients with advanced solid tumors [6]. Structural work has also helped explain how LAT1 recognizes amino acids and drug-like ligands, which supports the design of inhibitors with useful potency and selectivity profiles [7,22].
Another notable target is xCT, or SLC7A11, the cystine-glutamate antiporter. xCT supports glutathione synthesis, redox control, and resistance to oxidative stress. In several tumor settings, that makes it a biologically relevant survival factor. In non-small cell lung cancer, xCT-mediated metabolic changes have been linked to disease progression [8]. These examples show that transporter targets in oncology are not only biologically interesting but increasingly actionable [6-8].
ABC transporters are relevant in oncology for a different reason. Proteins such as ABCB1/P-glycoprotein, MRP1, and MRP4 can reduce intracellular drug exposure and contribute to multidrug resistance. Recent structural studies have improved understanding of how these transporters recognize substrates and inhibitors, which should support more informed medicinal chemistry in future programs [9-11].
Why transporters are difficult to screen and optimize
Strong biology does not make transporters easy targets. In practice, these programs remain challenging because of membrane dependence, conformational flexibility, assay complexity, and selectivity risk.
Membrane context and conformational dynamics
Transporters are membrane proteins, and many behave differently once removed from their native setting. Lipids, ions, membrane potential, glycosylation, and partner proteins can all influence function and ligand recognition [7,9-11]. Some transporters can be purified successfully and studied in detail. Others become less representative of the biologically relevant target state once taken out of the membrane.
Conformational dynamics add a second layer of difficulty. Transporters generally cycle through outward-open, occluded, inward-open, and ligand-stabilized states. As a result, the best compound is not always the one that binds most tightly in a static assay. A useful ligand may need to favor a specific state, slow the transport cycle, or prevent the transporter from adopting a productive conformation [1,3,7]. That is one reason transporter discovery often depends on both functional data and binding-related data.
Assay design and selectivity challenges
Assay design is another common source of failure. A compound may appear to inhibit transport because it affects ion gradients, membrane integrity, trafficking, or cell health rather than because it binds the target directly. The opposite can also happen: a real binder may look weak in an early assay if the transporter is not presented in the right state.
Selectivity is often just as important. Many transporters belong to related families with similar folds or overlapping ligand preferences. A screening hit can therefore carry unwanted activity against close paralogs or anti-targets. This is especially important for efflux transporters, where broad inhibition may create major pharmacokinetic or safety problems [9-11].
For that reason, transporter programs usually need a combination of:
- a biologically relevant screening format
- rapid functional confirmation
- counterscreens against related transporters
- early off-platform or off-DNA confirmation
- disease-relevant cellular assays before large chemistry effort begins [1,7,9,15]
Uptake and efflux transporters require different discovery logic
It is useful to separate uptake transporters from efflux transporters because the discovery logic is often different.
Uptake transporters
Uptake transporters are most attractive when disease cells depend on imported nutrients, metabolites, or signaling molecules. This group includes LAT1, ASCT2, GLUT-family transporters, and several organic anion or cation transporters [1,6-8]. In cancer, blocking uptake can limit access to metabolites required for growth or redox balance. In neuroscience, uptake transporters can shape transmitter clearance and tissue exposure [1,3].
Common goals in uptake transporter programs include:
- blocking nutrient supply to tumor cells
- changing neurotransmitter clearance
- reducing uptake of harmful endogenous or exogenous molecules
- exploiting transporter expression for selective delivery
Efflux transporters
Efflux transporters often matter because they lower intracellular exposure to drugs or endogenous molecules. In tumors, this can contribute to multidrug resistance. In barrier tissues, it can limit distribution into protected compartments such as the brain. These programs usually place greater weight on selectivity because broad inhibition of efflux transport can cause unwanted systemic effects [9-11].
So while uptake and efflux transporters are both drug targets, they often require different assays, different pharmacological goals, and different medicinal chemistry strategies.
What a modern transporter discovery strategy should include
Transporter programs work best when the discovery plan is built around the biology of the target rather than around a default screening platform.
Start with target biology
The first step is to define the intended biological intervention as clearly as possible.
- Do you want to block nutrient import?
- Do you want to inhibit drug efflux?
- Do you want to prolong neurotransmitter signaling?
- Do you want to stabilize a specific conformational state?
- Do you want to use transporter expression for targeted uptake rather than inhibition?
These are different questions, and each points to a different kind of ligand and a different screening funnel [1,3,6,7].
Human biology should also shape target selection early. Transporters are especially suitable for this because disease relevance is often visible in genetics, tissue expression, metabolic dependency, or patient stratification markers [1,2]. When the biological rationale is strong, the technical cost of running a transporter campaign is much easier to justify.
Match the screening format to the target state
For transporters, target presentation is often as important as library design. A purified transporter in detergent, a target in nanodiscs, an overexpressed construct on cells, and an endogenous transporter in living cells do not provide the same biological information [7,12-15].
A strong strategy chooses the screening format that best matches the project hypothesis. If the transporter is stable and tractable after purification, a purified format may work well. If membrane context is central to the biology, then live-cell or membrane-based screening may be more informative from the start [7,12-16].
Confirm hits quickly with orthogonal assays
Transporter programs benefit from fast progression from broad screening to orthogonal validation. In practical terms, that means:
- confirm hits in functional uptake or efflux assays
- test selectivity against related transporters early
- resynthesize compounds off-DNA or off-platform quickly
- use structural or mutational data where available
- assess disease-relevant cellular phenotypes before investing heavily in chemistry [1,7,9,15]
The aim is not simply to collect hits. It is to identify compounds that can support a real mechanism and survive follow-up work.
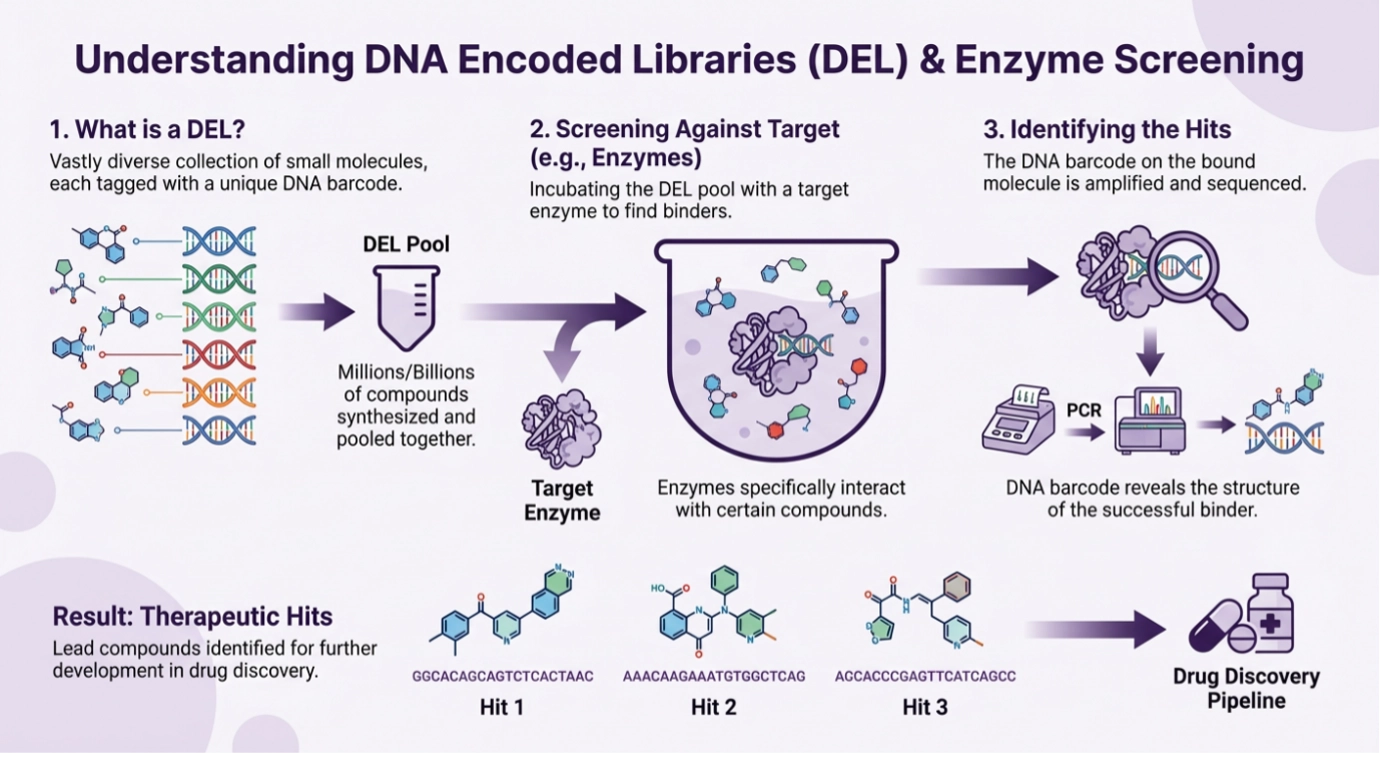
Why DNA-encoded libraries are useful for transporter targets
DNA-encoded library screening is particularly relevant for transporters because these targets often need both broad chemical diversity and a screening format that preserves biology.
Broad chemical diversity
DEL gives discovery teams access to very large chemical collections. That matters for transporter targets because many remain underexplored, and known ligand matter can be limited [12-16]. Broad chemical sampling improves the chance of finding differentiated starting points, especially when conventional screening has produced narrow or low-quality chemistry.
Live-cell and membrane-protein screening formats
Historically, DEL was mainly associated with purified proteins. Over time, that has changed. Several studies now show that DEL can be applied to membrane proteins and live-cell settings [12-16].
Wu and colleagues showed that cell-based DEL selection could identify antagonists of the NK3 tachykinin receptor on cells [12]. Cai and colleagues extended DEL selection to proteins on and within living cells [13]. Petersen and colleagues reported screening a multimillion-member DNA-encoded library inside a living cell using Xenopus oocytes [14]. Huang and colleagues later showed DEL selection against endogenous membrane proteins on live cells without requiring overexpression or genetic engineering [15]. More recently, live-cell DEL has also been used for agonist discovery on membrane proteins [16].
For transporter targets, these studies are important because many transporters are difficult to purify or maintain in a useful state outside cells. When target presentation is the bottleneck, live-cell or membrane-context DEL can offer a more practical route to relevant ligand discovery [12-16].
Why this matters for transporter programs at Vipergen
Transporters are a good fit for discovery platforms that combine large-scale ligand discovery with biologically relevant target presentation. Vipergen’s published platform information describes DNA-encoded library screening in living cells, workflows for purified proteins and integral membrane proteins, and support for difficult targets where conventional formats are limiting [17-20].
DEL screening for difficult membrane proteins
That positioning is directly relevant to transporter targets. Many transporters are difficult because purified protein is unstable, the target is strongly state-dependent, or membrane context is needed for meaningful pharmacology. A platform built to work with difficult membrane proteins can therefore be a better starting point than a standard screening workflow designed mainly around soluble targets [17-20].
Live-cell workflows and follow-up chemistry
Vipergen also describes the follow-up capabilities that matter in real discovery work, including hit triage, off-DNA resynthesis, medicinal chemistry support, and selectivity-oriented workflows [17,18,21]. For transporter programs, that combination is important. The key question is rarely whether a platform can generate an enrichment signal. The real question is whether it can move from that signal to confirmed compounds with a plausible mechanism and a workable validation path.
For teams working on transporters, that makes the overall workflow more important than any single technology label. Screening scale, target context, and rapid follow-up need to work together. Based on Vipergen’s published materials, that is exactly the kind of problem the platform is designed to address [17-21].
Conclusion
Transporters are now one of the more useful and still underexplored target classes in drug discovery. The biology is strong, the clinical precedent is real, and the technical tools are improving. What still makes these programs difficult is the need to screen the right target state, use the right assay logic, and move quickly from hits to mechanism [1-16,22].
That is why discovery strategy matters so much here. Broad chemistry helps, but only if the screen reflects the biology that matters. Structural insight helps, but only if it connects to function. Screening scale helps, but only if it leads to compounds that stand up in follow-up work.
If your transporter program has been slowed by target purification, weak translation from biochemical assays, or a lack of differentiated chemistry, it makes sense to use a platform built for difficult membrane proteins and native-context screening. Vipergen combines large-scale DNA-encoded library discovery with living-cell and membrane-protein workflows, followed by rapid hit confirmation and medicinal chemistry support. For transporter targets, that can shorten the path from target hypothesis to validated hits. Contact Vipergen to discuss your transporter target and the best route to a screening strategy that fits the biology [17-21].
References
[1] Lin L., Yee S.W., Kim R.B., Giacomini K.M. SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov (2015), 14, 543-560. DOI: 10.1038/nrd4626.
[2] César-Razquin A., Snijder B., Frappier-Brinton T., Isserlin R., Gyimesi G., Bai X., et al. A Call for Systematic Research on Solute Carriers. Cell (2015), 162(3), 478-487. DOI: 10.1016/j.cell.2015.07.022.
[3] Coleman J.A., Green E.M., Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature (2016), 532, 334-339. DOI: 10.1038/nature17629.
[4] Zinman B., Wanner C., Lachin J.M., Fitchett D., Bluhmki E., Hantel S., et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med (2015), 373, 2117-2128. DOI: 10.1056/NEJMoa1504720.
[5] Perkovic V., Jardine M.J., Neal B., Bompoint S., Heerspink H.J.L., Charytan D.M., et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med (2019), 380, 2295-2306. DOI: 10.1056/NEJMoa1811744.
[6] Okano N., Naruge D., Kawai K., Kobayashi T., Nagashima F., Endou H., et al. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Invest New Drugs (2020), 38, 1495-1506. DOI: 10.1007/s10637-020-00924-3.
[7] Lee Y., Wiriyasermkul P., Jin C., Quan L., Ohgaki R., Okuda S., et al. Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat Struct Mol Biol (2019), 26, 510-517. DOI: 10.1038/s41594-019-0237-7.
[8] Ji X., Qian J., Rahman S.M.J., Siska P.J., Zou Y., Harris B.K., et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene (2018), 37, 5007-5019. DOI: 10.1038/s41388-018-0307-z.
[9] Alam A., Kowal J., Broude E., Roninson I., Locher K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science (2019), 363(6428), 753-756. DOI: 10.1126/science.aav7102.
[10] Pietz H.L., Abbas A., Johnson Z.L., Oldham M.L., Suga H., Chen J. A macrocyclic peptide inhibitor traps MRP1 in a catalytically incompetent conformation. Proc Natl Acad Sci U S A (2023), 120(11), e2220012120. DOI: 10.1073/pnas.2220012120.
[11] Huang Y., Xue C., Wang L., Bu R., Mu J., Wang Y., et al. Structural basis for substrate and inhibitor recognition of human multidrug transporter MRP4. Commun Biol (2023), 6, 549. DOI: 10.1038/s42003-023-04935-7.
[12] Wu Z., Graybill T.L., Zeng X., Platchek M., Zhang J., Bodmer V.Q., et al. Cell-Based Selection Expands the Utility of DNA-Encoded Small-Molecule Library Technology to Cell Surface Drug Targets: Identification of Novel Antagonists of the NK3 Tachykinin Receptor. ACS Comb Sci (2015), 17(12), 722-731. DOI: 10.1021/acscombsci.5b00124.
[13] Cai B., Kim D., Akhand S.S., Sun Y., Cassell R.J., Alpsoy A., et al. Selection of DNA-Encoded Libraries to Protein Targets within and on Living Cells. J Am Chem Soc (2019), 141(43), 17057-17061. DOI: 10.1021/jacs.9b08085.
[14] Petersen L.K., Christensen A.B., Andersen J., Folkesson C.G., Kristensen O., Andersen C., et al. Screening of DNA-Encoded Small Molecule Libraries inside a Living Cell. J Am Chem Soc (2021), 143(7), 2751-2756. DOI: 10.1021/jacs.0c09213.
[15] Huang Y., Meng L., Nie Q., Zhou Y., Chen L., Yang S., et al. Selection of DNA-encoded chemical libraries against endogenous membrane proteins on live cells. Nat Chem (2021), 13, 77-88. DOI: 10.1038/s41557-020-00605-x.
[16] Huang Y., Hou R., Lam F.S., Jia Y., Zhou Y., He X., et al. Agonist Discovery for Membrane Proteins on Live Cells by Using DNA-encoded Libraries. J Am Chem Soc (2024), 146(35), 24638-24653. DOI: 10.1021/jacs.4c08624.
[17] Vipergen ApS. DNA-encoded library (DEL) | Vipergen – Get an early lead. Website. Accessed April 29, 2026.
[18] Vipergen ApS. High fidelity drug discovery – Get an early lead. Website. Accessed April 29, 2026.
[19] Vipergen ApS. DELs in Cells – Integral Membrane Proteins. Website. Accessed April 29, 2026.
[20] Vipergen ApS. Questions and Answers. Website. Accessed April 29, 2026.
[21] Vipergen ApS. Medicinal Chemistry Services for DNA-Encoded Library Screening. Website. Accessed April 29, 2026.
[22] Lee Y., Jin C., Ohgaki R., Xu M., Ogasawara S., Warshamanage R., et al. Structural basis of anticancer drug recognition and amino acid transport by LAT1. Nat Commun (2025), 16, 1635. DOI: 10.1038/s41467-025-56903-w.
Related Services
| Service | |
|---|---|
| Small molecule drug discovery for even hard-to-drug targets – identify inhibitors, binders and modulators | |
| Molecular Glue Direct | |
| PPI Inhibitor Direct | |
| Integral membrane proteins | |
| Specificity Direct – multiplexed screening of target and anti-targets | |
| Express – optimized for fast turn – around-time | |
| Snap – easy, fast, and affordable |