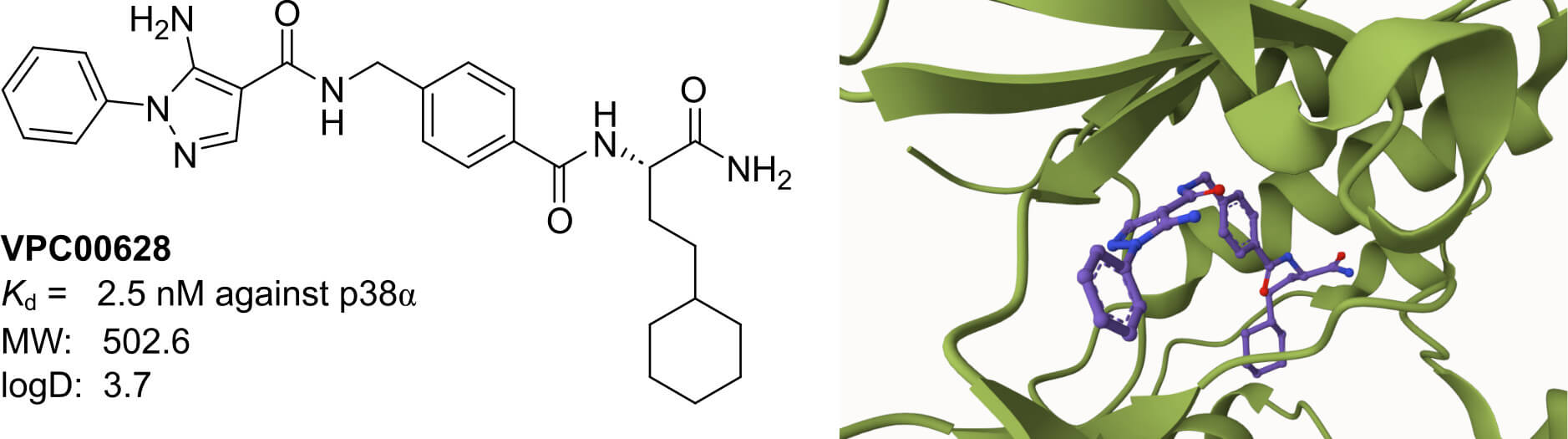
Medicinal Chemistry Services for DNA-Encoded Library Screening

Medicinal Chemistry Services for DNA-Encoded Library Screening
Introduction
In modern drug discovery, medicinal chemists have more data, more biological targets, and more modalities than ever before. Yet the path from primary screening to validated chemical hits remains a bottleneck for many R&D programs. High-throughput screening, fragment campaigns and DNA Encoded Libraries (DELs) can generate long lists of potential binders, but transforming those signals into experimentally confirmed hits – quickly, reproducible, and with a clear structure activity relationship – demands specialized expertise.
Medicinal chemistry services support the discovery phase where early screening signals are converted into real, testable chemical matter and a clear decision path. For teams seeking outsourced medicinal chemistry support for early drug discovery, our DEL-focused approach delivers validated hit matter and a clear handoff into downstream optimization—whether that work happens in-house or with a medicinal chemistry CRO partner. In a typical workflow, medicinal chemistry helps teams: (1) identify and prioritize hit series, (2) confirm structures with synthesized compounds, and (3) build early structure–activity relationships (SAR) that guide the next optimization steps.
Hit identification can originate from several discovery approaches—such as high-throughput screening (HTS), fragment-based discovery (FBDD), structure-based design (SBDD), or DNA-encoded libraries (DELs). DEL is especially valuable when you want to explore very large chemical space efficiently and quickly translate selection signals into prioritized chemical series for follow-up. On this page, Vipergen’s medicinal chemistry contribution is described with a focus on DEL-enabled hit identification and rapid off-DNA resynthesis/validation—so you can move from sequencing output to decision-ready compounds without losing momentum.
Vipergen was founded to provide exactly that service as a medicinal chemistry company whose platform is built around the synthesis and screening of DELs. We focus on the initial part of the drug discovery pipeline (figure 1) and can take your target protein of interest (POI) to early lead like compounds in a few months. Hereby Vipergen’s medicinal chemistry platform can help to deliver the greatest impact and the highest return of investment. By concentrating our resources on DEL construction, selections, data analysis, and hit re-synthesis, we offer a streamlined medicinal chemistry service that plugs directly into your discovery engine – no matter whether your downstream chemistry is handled in-house or by another medicinal chemistry CRO.
Quick definitions: DEL = DNA-encoded library; SAR = structure–activity relationship; NGS = next-generation sequencing; FBDD/SBDD = fragment-/structure-based drug discovery; PPI = protein–protein interaction.
Figure 1: Overview of the drug discovery and development process. Vipergen’s medicinal chemistry services support hit identification and lead identification by combining DEL screening, sequencing/informatics analysis, series nomination, and rapid off-DNA resynthesis—enabling faster confirmation and cleaner handoff into downstream hit-to-lead activities.
Throughout this article we will explain how Vipergen’s focused medicinal chemistry solutions accelerate early drug discovery while maintaining full traceability, uncompromising quality, and competitive turnaround times. We deliver high-quality DEL hits which can readily be resynthesized off-DNA ready for the next phase.
Ready to evaluate DEL for your target? Contact us to discuss target format, selection strategy, and the fastest path to off-DNA validation.
What are Medicinal Chemistry Services?
Medicinal chemistry services help transform early screening signals into validated, optimizable chemical matter. In practice, this means designing and synthesizing compounds, confirming hit structures, building structure–activity relationships (SAR), and improving potency, selectivity, and developability so programs can progress from hit → lead efficiently.
Medicinal chemists work at the interface of biology and chemistry: they interpret assay data, propose hypotheses for binding and selectivity, and rapidly test those hypotheses through iterative synthesis and profiling. The goal is not simply “more compounds,” but decision-ready series with clear SAR, quality data, and routes that scale.
Medicinal Chemistry Services at Vipergen
Vipergen supports your early drug discovery engine by combining DEL-enabled hit finding, data-driven triage, and rapid off-DNA resynthesis, so you can accelerate hit validation and make confident series decisions. While our platform is built around DNA-encoded libraries, our medicinal chemistry support is designed to plug into your pipeline—whether you run downstream chemistry in-house or with another CRO.
Typical outcomes we help deliver:
- Confirmed hit structures (off-DNA resynthesis + QC)
- Early SAR to prioritize series
- Selectivity strategies (target / anti-target thinking)
- Fast handoff into hit-to-lead and lead optimization
Key Service Offerings
Vipergen’s medicinal chemistry support is purpose-built for DNA-Encoded Library (DEL) hit identification and rapid off-DNA resynthesis—so you can move from selection signals to confirmable, decision-ready chemical matter with confidence. We combine DEL platform execution, sequencing/informatics, and follow-up chemistry to deliver validated hits and clear next-step recommendations for your program.
Common challenge: Early DEL campaigns can generate compelling enrichment signals, but teams often lose time deciding what to trust, what to resynthesize first, and how to translate barcode counts into experimentally confirmed hits.
Vipergen’s solution: We provide a focused medicinal chemistry service built around DEL hit identification, chemically informed triage, and rapid off-DNA resynthesis—so you can move from sequencing output to decision-ready compounds quickly and confidently.
Request a consultation: Share your target, desired selectivity profile, and preferred confirmation assays—we’ll propose a screening + resynthesis plan sized to your decision points.
1. DEL Screening for Hit Identification
What it is: We run DEL screening to identify binders against your target and generate a ranked, interpretable hit list. DEL is especially useful when you need access to broad chemical space and want early insights into binding motifs and SAR trends.
What you get:
- DEL selection execution aligned to your target biology (typical workflows include selection design, conditions optimization, and selection rounds as needed)
- Sequencing + informatics analysis to identify enriched features, clusters, and priority series
- Hit list with context (enrichment, clustering/series calls, and recommended shortlist for resynthesis)
- Project-ready reporting package that supports downstream validation and decision-making
When this fits best:
- You need novel chemotypes quickly
- You want SAR-rich starting points from library-level structure patterns
- You need a hit-finding method that complements other discovery approaches
How to engage (examples)
- Fixed-scope DEL selection + report (single target)
- DEL selection with iterative data reviews and shortlist refinement before resynthesis
- DEL selection plus add-on conditions (e.g., competition conditions or selectivity strategy, when applicable)
2. Hit Triage, Series Nomination & Next-Step Recommendations
DEL outputs are most valuable when they’re translated into a clear, defensible plan. We help you prioritize what to resynthesize first and how to validate efficiently.
What you get:
- Series nomination based on clustering and chemical logic (not just top enrichment)
- Shortlist strategy (e.g., diversify across series, prioritize tractable chemistry, balance novelty vs. risk)
- Validation roadmap outlining what to confirm first and what data to generate next (for your internal team or preferred CRO)
How to engage (examples)
- Quick-turn shortlist + recommendations after sequencing
- Joint review sessions to align on series priorities and validation sequencing
3. Off-DNA Hit Resynthesis & Quality Control
What it is: We resynthesize prioritized DEL-derived hits off DNA to generate clean, fully characterized compounds for confirmation work.
What you get
- Off-DNA resynthesis of selected hits/representatives from nominated series in mg quantities
- Purification and QC so compounds are suitable for downstream validation
- Compound delivery prepared for your biochemical/biophysical/cellular confirmation assays
Why it matters: DEL selections can generate strong signals—but program decisions require real compounds. Resynthesis closes the loop, letting you confirm binding and prioritize series with confidence.
How to engage (examples)
- Resynthesize a focused shortlist (fast confirmation set)
- Resynthesize representatives across multiple series to de-risk and compare options
What we don’t do (and how we support you anyway)
Vipergen focuses on DEL-enabled hit identification and off-DNA resynthesis. For broader medicinal chemistry (hit-to-lead and lead optimization), we equip your internal team or CRO partner with a validated starting set, series rationale, and a handoff package that shortens downstream cycles.
Our deliverables are designed to accelerate downstream hit-to-lead and lead optimization by starting those phases with confirmed structures, prioritized series, and early SAR signals—not raw sequencing output.
Why Choose Vipergen for Medicinal Chemistry?
- Traceability + quality: high-fidelity library build and clean handoff to off-DNA compounds
- Integrated wet + dry: you explicitly call out sequencing-file analysis + informatics pipelines
- Speed to confirmable matter: resynthesis capability + in-house building blocks
- Unique platform strengths: YoctoReactor (Figure 2)/BTE/cBTE as technology differentiators
- IP/series handling: your “hit reservation and series nomination” concept is a strong trust-builder—bring it up here too
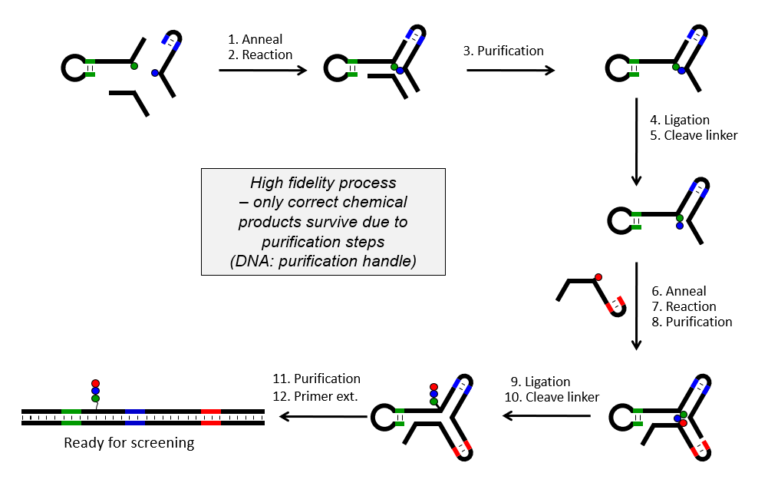
Figure 2: Vipergen’s method for DEL synthesis utilizing the YoctoReactor. The YoctoReactor ensures high fidelity libraries without truncates as only correct chemical products survive due to purification step.
Why utilizing DEL screening as part of medicinal chemistry campaign?
-
- It Expands Chemical Space Exploration: Where commercial screening decks cover up to one million compounds, a well-designed DEL explores hundreds of millions of small molecules. This jump of almost three-fold in magnitude uncovers novel scaffolds and binding modes which traditional small-molecule screening libraries rarely touch. Vipergen’s DELs introduces thousands of designer building blocks allowing for full exploration of chemical space. Our team works daily to expand the chemical space our DNA encoded libraries explores.
- It Demands Precise Synthesis: Building a high-quality multi-million-member DEL is impossible without high-fidelity chemistry. At Vipergen we utilize our proprietary YoctoReactor technology (Figure 2) to construct DELs with a complete match between DNA barcode and the displayed small molecule.
- It Requires Bioinformatics as Much as Bench Work: DEL selection data arrives as sequencing files, not IC50-values. Turning barcode counts into a ranked list of real molecules calls for dedicated informatics pipelines, sophisticated enrichment algorithms, and chemist who can understand both the statistics and the synthetic feasibility behind each hit. Coupling wet-lab and dry-lab expertise in one team minimizes hands-offs and maximizes interpretability.
- It gives access to maximum diversity: Vipergen’s DELs are optimized to maximize 3D diversity by introducing building blocks which display different spatial orientations. This is done by introducing chemical building blocks with distinct cores to deliver 3D diversity. To further enhance 3D diversity these cores are diversified by their stereo- and positional-isomers.
- It Gives Access to Early Structure Activity Relationship: Vipergen’s DELs includes core scaffolds decorated with diverse sets of substituents. This means that once a hit is identified, structure activity relationship (SAR) information is readily available from the DEL screen.
- It Expands Chemical Space Exploration: Where commercial screening decks cover up to one million compounds, a well-designed DEL explores hundreds of millions of small molecules. This jump of almost three-fold in magnitude uncovers novel scaffolds and binding modes which traditional small-molecule screening libraries rarely touch. Vipergen’s DELs introduces thousands of designer building blocks allowing for full exploration of chemical space. Our team works daily to expand the chemical space our DNA encoded libraries explores.
Technology Overview behind Vipergen’s DEL-enabled medicinal chemistry
Vipergen’s platform combines high-fidelity DEL construction, flexible selection formats, and data-to-compound follow-up chemistry:
- YoctoReactor® (yR) DEL synthesis: Enables construction of very large, drug-like DELs with high fidelity between barcode and small molecule.
- Binder Trap Enrichment® (BTE): Homogeneous, solution-phase selection workflows designed to avoid immobilization and matrix effects common in solid-phase approaches.
- Cellular Binder Trap Enrichment® (cBTE): Enables DEL screening inside living cells, supporting target classes where purified protein is challenging and improving physiological relevance (Figure 3).
- NGS + informatics analysis: Turns sequencing output into ranked hit lists, clusters, and chemically actionable series shortlists.
- Off-DNA resynthesis + QC: Converts prioritized sequences into purified, QC-verified compounds ready for confirmation assays and next-step decision making.
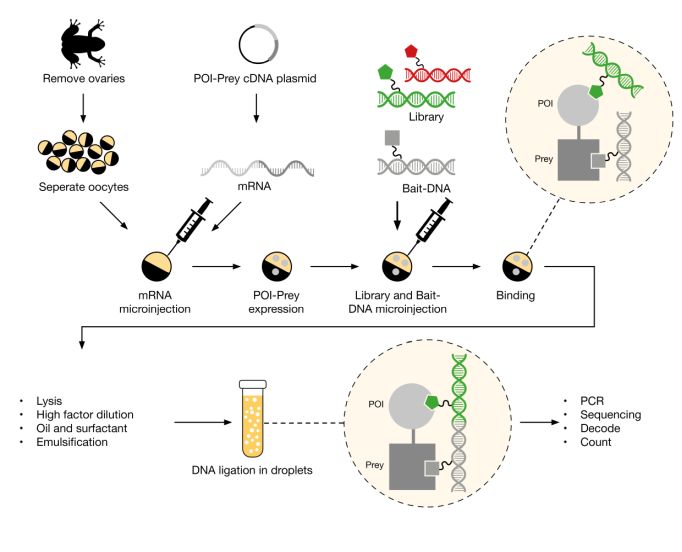
Figure 3: Overview of Vipergen’s cellular Binder Trap Enrichment technology.
Targets & applications supported by Vipergen DEL screening
Vipergen’s DEL screening formats are designed to support challenging discovery scenarios, including:
- Membrane and hard-to-purify targets using in-cell screening approaches where purified protein is not required.
- Protein–protein interaction (PPI) inhibitor discovery and related challenging interfaces.
- Molecular glue discovery using multiplexed screening strategies where applicable.
- Selectivity-focused screening including counter-screens against anti-targets to prioritize series with cleaner profiles early.
- In vitro DEL screening as an alternative when a purified target format is preferred.
Library Architecture
- Scaffold-Centric Diversity: Each library member contains three-dimensional and sp3-rich core scaffolds chosen for synthetic tractability and physicochemical balance (Average for Vipergen’s Lib56: Fsp3 = 0.6; cLogP = 0.75; HBA/HBD within Lipinski limits; 343 different core-scaffolds).
- Orthogonal Encoding Tags: Our YoctoReactor technology ensures complete eliminations of truncates, ensuring every compound is uniquely addressable.
Selection Workflow
- Screening in Living Cells: Our unique screening technique inside living cells allows for rapid screening against most target classes including membrane targets as purified protein is not needed. Read more about cellular Binder Trap Enrichment here.
- Screening in vitro: Besides screening in cells, we also perform more traditional DEL screenings in vitro. Here we utilize our Binder Trap Enrichment, which omits the need for target immobilization.
- Positive and Negative Selection Rounds: We can directly screen both against target protein, but also counter-screen against non-targets which you want selectivity against.
- Screening Beyond Inhibitors: We have developed multiplex screening technologies, which allow for the direct identification of both molecular glues and protein-protein interaction inhibitors. Check out our full list of services here.
Analysis Workflow
- Next-Generation Sequencing (NGS) and Analysis: We analyze NGS data and compile a hit list and report. This includes metrics from the screen and chemical structure information.
- Hit reservation and Chemical Series Nomination: Hits are reserved for our clients for 1 year, where you may resynthesize and test in relevant assays. In this time, you may nominate chemical series, which becomes exclusive for you.
Hit Resynthesis: Converting Sequences into Samples
Waiting several months to obtain off-DNA hits negates the speed advantage of DEL Screening. We have synthetic capabilities to speed up that timeline as we have almost all building blocks in-house and a dedicated team ready to synthesize hit compounds at mg-scale which can fit into your medicinal chemistry campaign. As our DELs are constructed stepwise, resynthesis generally follows the same synthetic transformations making route planning directly available. All hit compounds are purified and undergo quality control. For most projects, the first milligrams of finalized off-DNA hits ship within a month.
Need validated hits fast? Ask about typical resynthesis set sizes and a timeline aligned to your next program gate.
Common challenge: DEL screening is fast, but if off-DNA compounds arrive months later, the speed advantage disappears and programs stall at the validation step.
Vipergen’s solution: We convert prioritized DEL hits into purified, QC-verified off-DNA compounds on a competitive timeline—so your team can confirm activity and prioritize series without delay.
Benefits of outsourcing DEL-enabled hit identification and resynthesis
Partnering with a DEL specialist can reduce time-to-decision and de-risk early discovery by combining platform access with focused medicinal chemistry execution:
- Acceleration: Move from selection data to validated compounds quickly, preserving the speed advantage of DEL screening.
- Reduced false starts: High-fidelity library design and chemically informed triage help prioritize series that are resynthesizable and testable.
- Integrated delivery: One team covers DEL execution, sequencing analysis, and follow-up resynthesis—minimizing handoffs and interpretation gaps.
- Flexibility: Engage in fee-for-service screening, selectivity add-ons, or multiplex strategies depending on your target biology and decision points.
Quality, Traceability, and Confidentiality
Vipergen’s DEL-enabled medicinal chemistry services are built around traceability and fit-for-purpose quality control—from library design through hit nomination and off-DNA resynthesis.
How we support quality and data integrity
- Traceable data flow from selection output (sequencing files) through analysis and reporting
- Purification and QC of off-DNA hits prior to shipment to support reliable confirmation testing
- Chemically informed triage that accounts for synthetic feasibility and series logic, not enrichment alone
We routinely work under standard confidentiality terms (e.g., CDA/NDA) and align deliverables and reporting formats to your internal documentation needs.
Engagement Models that Respect Your Pipeline
Our DEL screening services can be scaled based on your needs. We can e.g. dimension our medicinal chemistry services to:
- Easy and fast DEL screening in cells, where we initially perform an expression study, which is followed by one round of screening.
- Direct selectivity screens, where we expand the screening against anti-target protein(s).
- Multiplex screening to identify molecular glues and protein-protein interaction inhibitors.
Common challenge: Discovery teams have different constraints—target format, selectivity requirements, timelines, and internal capacity—so a one-size-fits-all engagement slows decisions.
Vipergen’s solution: Our medicinal chemistry services are modular: you can start lean and expand scope only when the data justify it.
Check out the full series of services here and contact us about your project.
Related Services
| Service | |
|---|---|
| Small molecule drug discovery for even hard-to-drug targets – identify inhibitors, binders and modulators | |
| Molecular Glue Direct | |
| PPI Inhibitor Direct | |
| Integral membrane proteins | |
| Specificity Direct – multiplexed screening of target and anti-targets | |
| Express – optimized for fast turn – around-time | |
| Snap – easy, fast, and affordable |