


Novel inhibitors of an inflammatory kinase – found first try
Using Binder Trap Enrichment screening against a 12.6 million-compound yoctoReactor library, Vipergen identified selective, single-digit nanomolar inhibitors of the p38α MAP kinase in a single round of selection. X-ray crystal structures confirmed a previously undescribed binding mode
7 nM
1
12.6M
92%
A well-validated target that conventional approaches had failed to crack
p38α MAP kinase (MAPK14) is a central regulator of pro-inflammatory cytokines (including TNF-α, IL-1, and IL-6) and attracted decades of drug discovery investment for inflammatory diseases. Every clinical program ultimately failed, not for lack of potent inhibitors, but for reasons rooted in the target’s biology: compensatory upregulation of parallel MAPK pathways causes cytokine levels to rebound in hepatocytes and cardiomyocytes produces mechanism-based toxicity that more selective compounds cannot escape.
p38α therefore remains a valuable benchmark target being well-characterized, with abundant structural data and robust assays even as its therapeutic utility as narrowed. What the field lacked was not better inhibitors per se, but genuinely novel chemical matter to probe its biology from new angles. Existing series shared common hinge-binding motifs, a naïve, unbiased screen offered the prospect of something structurally distinct.
One tube. One round. 12.6 million compounds
Vipergen screened p38α against Lib022, a yoctoReactor-generated DNA-encoded library (DEL) comprising 12.6 million compounds, each uniquely identified by its covalently attached DNA barcode. Crucially, the library was not designed with kinases in mind: no prior structural knowledge of the target was incorporated into its design, making this a genuine test of the platform’s ability to discover novel chemical matter from scratch.
Screening was performed using Binder Trap Enrichment (BTE), Vipergen’s proprietary homogeneous solution-phase selection technology. Rather than immobilizing the target to a solid support, which risks denaturation, matrix binding artefacts, and low signal-to-noise, BTE traps target–ligand complexes within individual water-in-oil emulsion droplets.
-
Equilibrium binding
p38α, conjugated to a short DNA tag, was incubated with Lib022 in solution, allowing genuine equilibrium binding to occur. -
Dissociation & emulsification
The mixture was diluted to trigger dissociation of non-binders, then partitioned into billions of water-in-oil droplets. Binders, co-trapped with their target protein, remain together; non-binders are distributed randomly across the excess of empty droplets. -
Ligation & readout
Within each droplet, target DNA and library DNA are ligated, recording the binding event. After emulsion breakage, ligation products are PCR-amplified, sequenced, and counted. Compounds appearing at high frequency are binders; random co-trapping produces only low-count background. -
Hit decoding & resynthesis
DNA sequencing of ~500,000 reads yielded 236 primary hits. A Tanimoto similarity analysis clustered the top 97 into four distinct series. Twenty-four representative compounds were resynthesized off-DNA for biochemical and cellular validation.
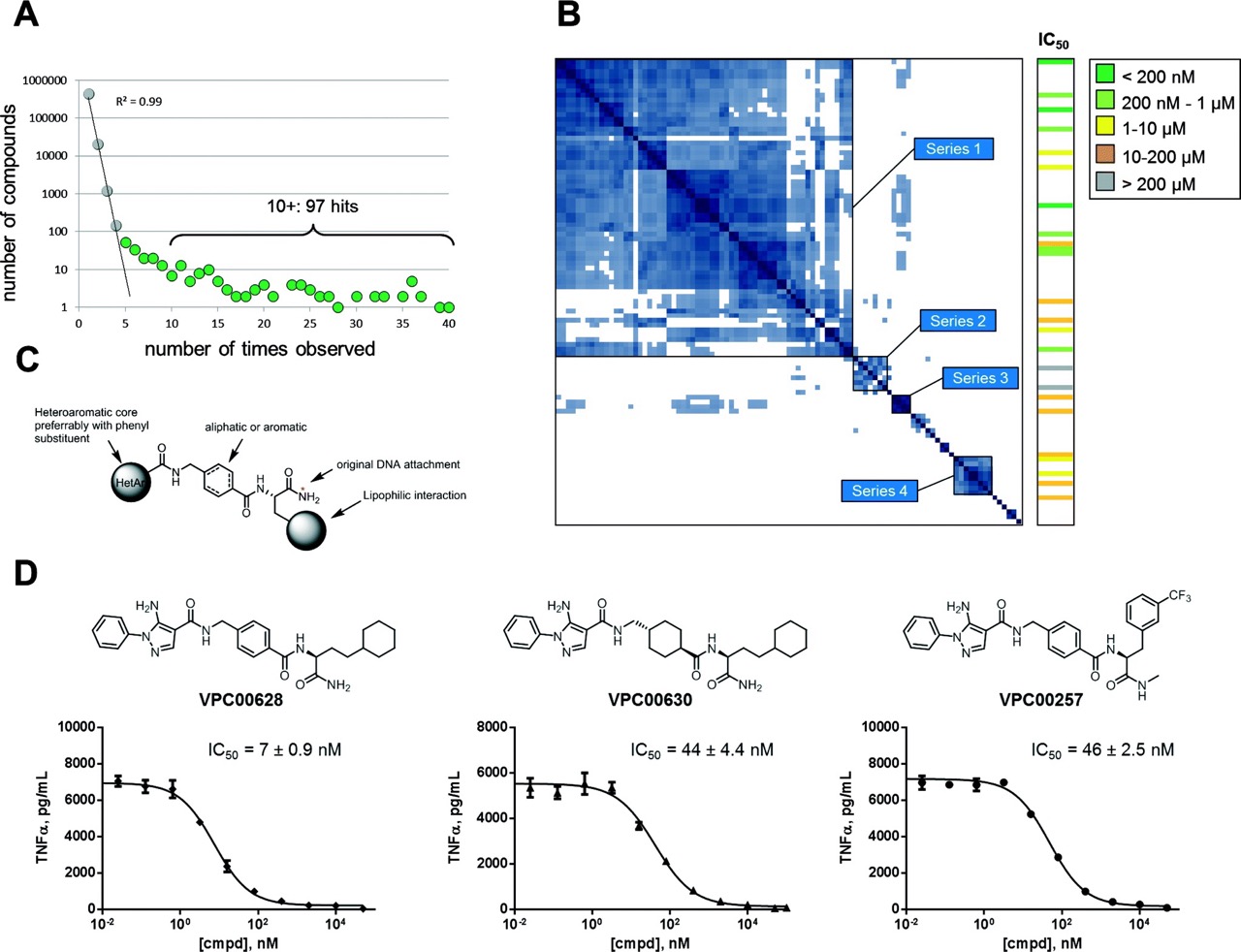
Figure 1.
A) BBTE signal plot showing the distribution of sequence counts; hits (green) separate
cleanly from random background (grey) above threshold of five observations. B) Tanimoto similarity revealing four distinct hit series. C) Generalized structure of observed hits. D) Structures and dose-response curves of three potent inhibitors in cellular TNF-α secretion assay.
Let’s discuss your next discovery program.
From enzymes to protein – protein interactions, transcription factors and epigenetic readers, Vipergen’s platform is built to deliver genuine hits – fast.
Potent, selective inhibitors and a novel binding mode
Of 24 resynthesized compounds, 22 were confirmed as p38α inhibitors yielding a hit confirmation rate of 92%, underlying the low false-positive rate inherent to BTE. The primary hit series yielded inhibitors spanning from low micromolar to single-digit nanomolar potency in biochemical assays, with lead compound VPC00628 achieving a cellular IC50 of 7 nM in a TNF-α secretion assay in human THP-1 monocytic cells.
7 nM
22/24
4 series
p38α/β only
Critically, the structural novelty of the hits went beyond selectivity numbers. X-ray crystallography of the p38α-VPC00628 complex at 1.5 Å resolution revealed a canonical type-II (‘DFG-out’) binding mode, yet one achieved through a strikingly atypical set of interactions. The compounds lack the hinge-binding motifs considered hallmarks of kinase inhibitors, instead forming a hydrogen bond-network via its pyrazole nitrogen with the backbone M109, while a cyclohexane decoration occupies the hydrophobic pocket opened by the DFG-out conformation.
Most remarkably, VPC00628 induced a pronounced distortion of the P-loop, a structural alteration not previously observed in complexes with established type-II inhibitors such as BIRB796 or sorafenib. This generated a channel that, in the original DNA-encoded compound, accommodated the linker to the DNA tag itself. This is a clear illustration of how DEL-derived hits can encode structural information about the screening format directly into their binding geometry.
Structural Insight
The P-loop distortion observed in the VPC00628 crystal structure is distinct from all other known p38α type-II inhibitor complexes. It opens a previously underexploited pocket between the P-loop and αC helix — a tractable vector for future medicinal chemistry and PROTAC discovery.
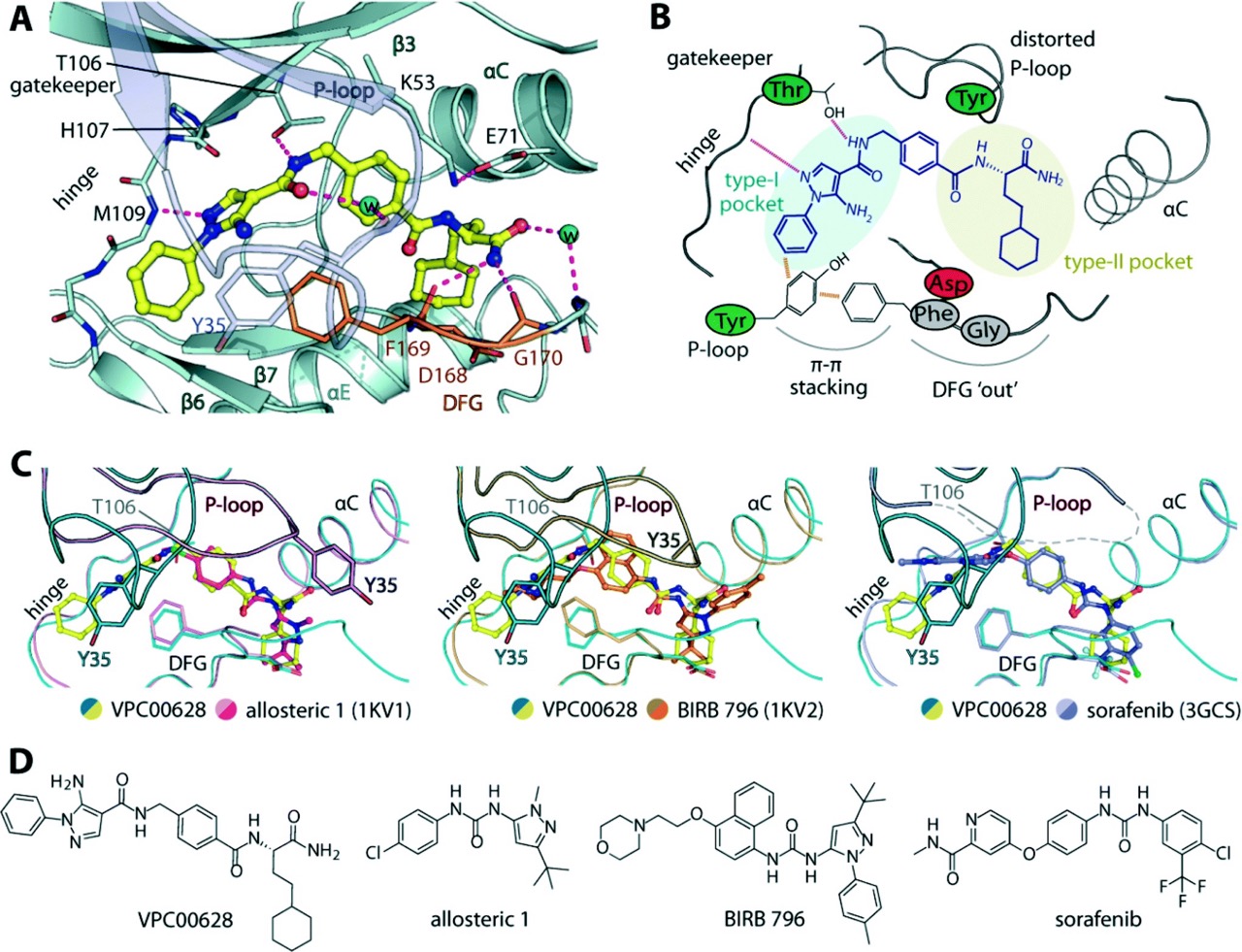
Figure 2.
Crystal structure of p38α in complex with VPC00628 (PDB: 5LAR), refined at 1.5 Å. A) Detailed interaction of the inhibitor with the kinase, with hydrogen bonds (magenta dashed lines) and water molecules (cyan spheres). B) Schematic illustration of the key interactions and structural alterations upon accommodation of the inhibitor within the kinase type-I and type-II pockets.
Truly naïve discovery without prior knowledge required
This study is perhaps the clearest demonstration of what BTE can deliver when given an unconstrained library and a well-defined target. No kinase-biased building blocks were incorporated. No kinase-specific pharmacophores were used as a starting point. The hit series emerged from chemistry selected on binding affinity alone, and yet yielded compounds potent enough for cellular validation, all without a single round of medicinal chemistry optimization.
The modular structure of the yoctoReactor library also means that follow-up chemistry is immediately tractable: each building block position can be varied independently, providing a direct path from screening hit to optimized lead.
Key takeaways for your program
No structural knowledge needed. The library’s built-in chemical diversity covers target classes from enzyme to protein-protein interactions, transcription factors and epigenetic readers without redesign.
Single-round selection is sufficient. BTE’s low false-positive rate means hits are genuine binders, not artefacts of repeated washes or matrix interactions.
Novel chemistry, not repurposed matter. BTE-derived hits routinely explore binding modes that fragment- and HTS-based approaches miss, as confirmed here by X-Ray crystallography
This case study is based on: Petersen et all., “Novel p38α MAP kinase inhibitors identified from yoctoReactor DNA-encoded small molecule library”, Med. Chem. Commun., 2016, 7, 1332-1339. DOI: 10.1039/c6md00241b. Open Access. Figures reproduced under the terms of the original CC BY-NC 3.0 license